Zhu Hengzhou, Ji Yi, Li Wenting, Wu Mianhua
First Clinical Medical College, Nanjing University of Traditional Chinese Medicine, Nanjing, Jiangsu 210000, P.R. China.
Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine Prevention and Treatment of Tumor, Institute of Oncology, The First Clinical Medical College, Nanjing, Jiangsu 210000, P.R. China.
Oncol Lett. 2019 Oct;18(4):3778-3786. doi: 10.3892/ol.2019.10698. Epub 2019 Aug 1.
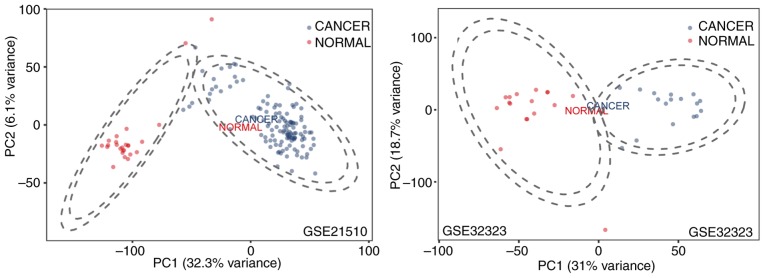
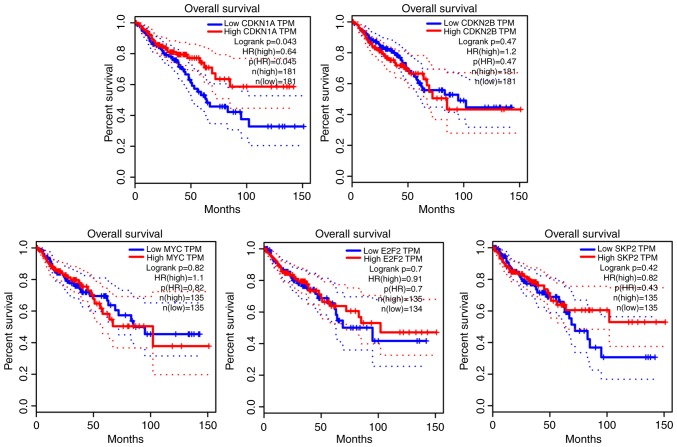
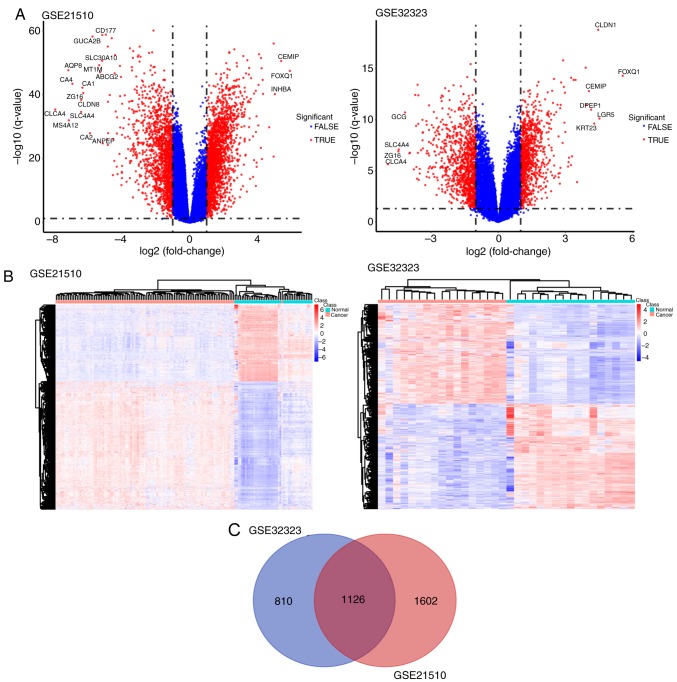
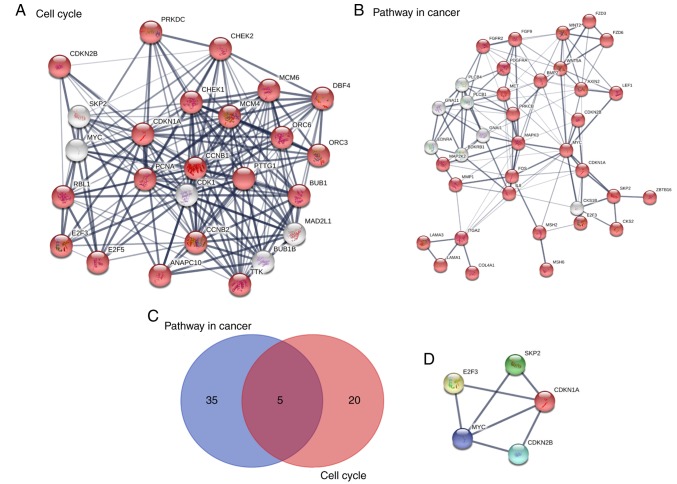
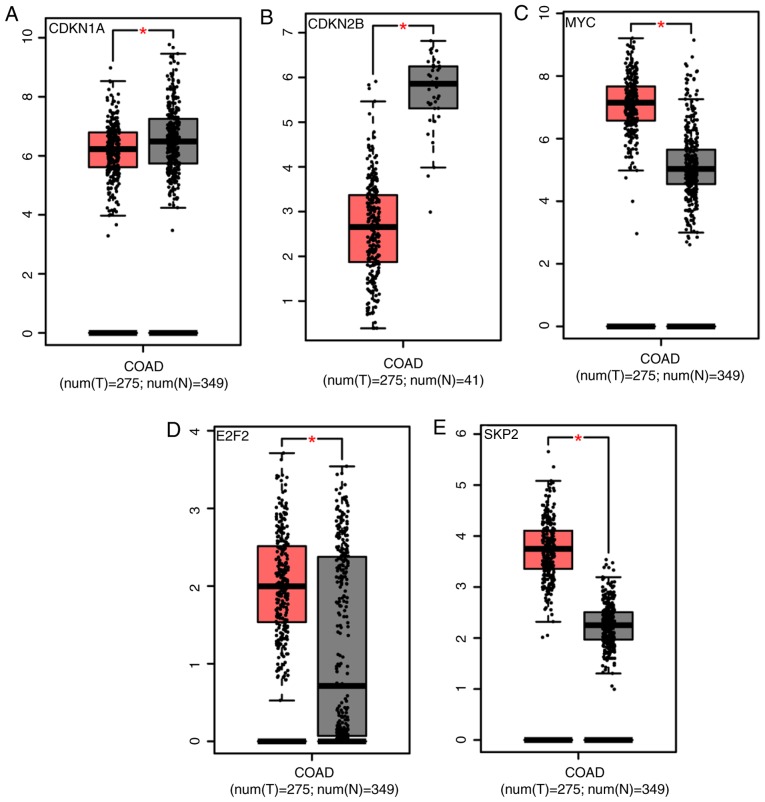
The aim of the present study was to identify key genes in colorectal cancer (CRC) that could be used to reliably diagnose this disease and to explore the potential underlying mechanisms . The gene expression profiles of primary human cancer datasets GSE21510 and GSE32323 were downloaded from the Gene Expression Omnibus database. The limma R software package was used to identify differentially expressed (DE) genes. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed on DE genes using the Database for Annotation, Visualization and Integrated Discovery. The Search Tool for the Retrieval of Interacting Genes/Proteins database was used to construct a protein-protein interaction (PPI) network of the DE genes. Survival rate was analyzed and visualized using The Cancer Genome Atlas (TCGA). A total of 1,126 genes were significantly DE in the present study. All DE genes were enriched in KEGG pathways including 'cell cycle', 'mineral absorption', 'pancreatic secretion', 'pathways in cancer', 'metabolic pathways', 'aldosterone-regulated sodium reabsorption' and 'Wnt signaling pathway'. A total of 5 hub genes enriched in cell cycle and tumor-associated pathways, including E2F2, SKP2, MYC, CDKN1A and CDKN2B, were significantly DE and validated between tumor and normal tissues. CDKN1A and CDKN2B were identified within the PPI network using the Molecular Complex Detection algorithm. Survival and content distribution analyses of 362 clinical samples from TCGA revealed that CDKN1A effectively predicted the prognosis of patients. The present study identified key genes and potential signaling pathways involved in CRC. These findings may provide new insights for survival assessment during the clinical diagnosis of CRC.
本研究的目的是鉴定可用于可靠诊断结直肠癌(CRC)的关键基因,并探索其潜在的机制。从基因表达综合数据库下载原发性人类癌症数据集GSE21510和GSE32323的基因表达谱。使用limma R软件包鉴定差异表达(DE)基因。使用注释、可视化和综合发现数据库对DE基因进行基因本体论和京都基因与基因组百科全书(KEGG)通路富集分析。使用检索相互作用基因/蛋白质数据库的搜索工具构建DE基因的蛋白质-蛋白质相互作用(PPI)网络。使用癌症基因组图谱(TCGA)分析并可视化生存率。在本研究中,共有1126个基因显著差异表达。所有DE基因均富集于KEGG通路,包括“细胞周期”、“矿物质吸收”、“胰腺分泌”、“癌症通路”、“代谢通路”、“醛固酮调节的钠重吸收”和“Wnt信号通路”。共有5个富集于细胞周期和肿瘤相关通路的枢纽基因,包括E2F2、SKP2、MYC、CDKN1A和CDKN2B,在肿瘤组织和正常组织之间显著差异表达并得到验证。使用分子复合物检测算法在PPI网络中鉴定出CDKN1A和CDKN2B。对来自TCGA的362份临床样本的生存和含量分布分析表明,CDKN1A可有效预测患者的预后。本研究鉴定了参与CRC的关键基因和潜在信号通路。这些发现可能为CRC临床诊断期间的生存评估提供新的见解。