School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, New South Wales, Australia.
Centre for Infectious Diseases and Microbiology - Public Health, Institute of Clinical Pathology and Medical Research, Westmead Hospital, New South Wales, Australia.
Microb Genom. 2021 Jun;7(6). doi: 10.1099/mgen.0.000310.
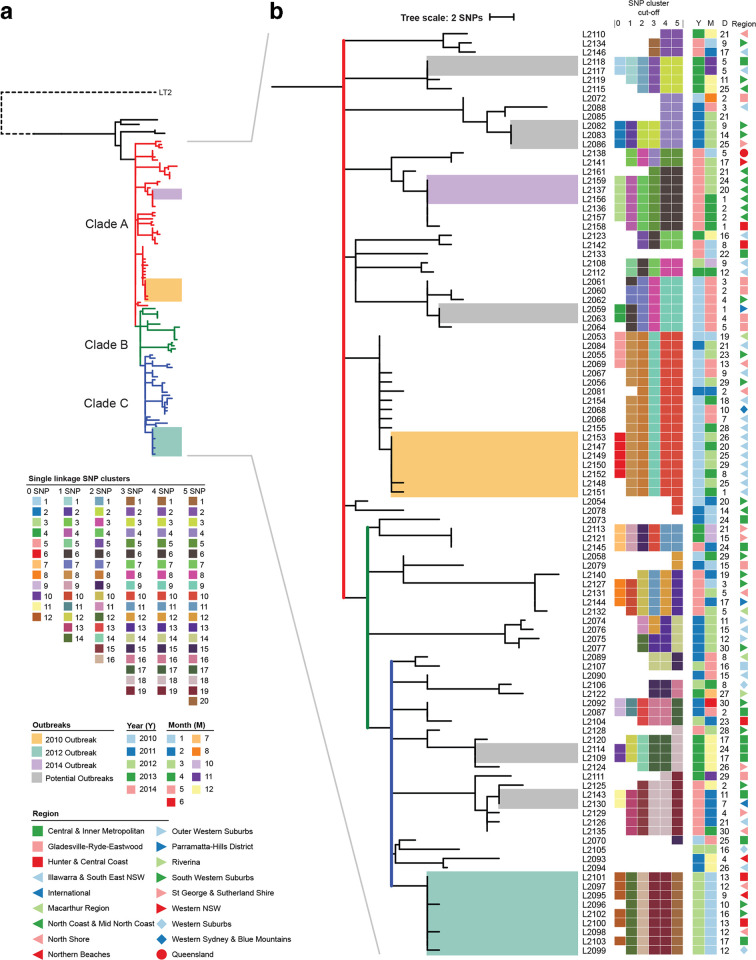
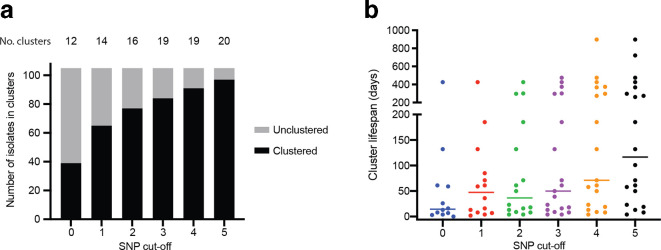
serovar Typhimurium is the leading cause of salmonellosis in Australia, and the ability to identify outbreaks and their sources is vital to public health. Here, we examined the utility of whole-genome sequencing (WGS), including complete genome sequencing with Oxford Nanopore technologies, in examining 105 isolates from an endemic multi-locus variable number tandem repeat analysis (MLVA) type over 5 years. The MLVA type was very homogeneous, with 90 % of the isolates falling into groups with a five SNP cut-off. We developed a new two-step approach for outbreak detection using WGS. The first clustering at a zero single nucleotide polymorphism (SNP) cut-off was used to detect outbreak clusters that each occurred within a 4 week window and then a second clustering with dynamically increased SNP cut-offs were used to generate outbreak investigation clusters capable of identifying all outbreak cases. This approach offered optimal specificity and sensitivity for outbreak detection and investigation, in particular of those caused by endemic MLVA types or clones with low genetic diversity. We further showed that inclusion of complete genome sequences detected no additional mutational events for genomic outbreak surveillance. Phylogenetic analysis found that the MLVA type was likely to have been derived recently from a single source that persisted over 5 years, and seeded numerous sporadic infections and outbreaks. Our findings suggest that SNP cut-offs for outbreak cluster detection and public-health surveillance should be based on the local diversity of the relevant strains over time. These findings have general applicability to outbreak detection of bacterial pathogens.
鼠伤寒血清型是澳大利亚沙门氏菌病的主要病因,识别暴发及其来源对于公共卫生至关重要。在这里,我们研究了全基因组测序(WGS)的实用性,包括使用牛津纳米孔技术进行完整基因组测序,以检查 5 年内 105 株来自地方性多位点可变数目串联重复分析(MLVA)类型的分离株。MLVA 类型非常同质,90%的分离株落入 5 个 SNP 截断的组中。我们开发了一种新的两步 WGS 暴发检测方法。首先,使用零单核苷酸多态性(SNP)截断值进行聚类,以检测在 4 周窗口内发生的暴发簇,然后使用动态增加 SNP 截断值的第二次聚类生成能够识别所有暴发病例的暴发调查簇。这种方法提供了最佳的特异性和敏感性,用于暴发检测和调查,特别是那些由地方性 MLVA 类型或遗传多样性低的克隆引起的暴发。我们进一步表明,包括完整的基因组序列并没有为基因组暴发监测检测到额外的突变事件。系统发育分析发现,MLVA 类型可能是由一个单一的来源在过去 5 年内持续存在,并引发了许多散发性感染和暴发。我们的研究结果表明,用于暴发簇检测和公共卫生监测的 SNP 截断值应基于相关菌株在时间上的本地多样性。这些发现对于细菌病原体的暴发检测具有普遍适用性。