Department of Drug Discovery & Biomedical Sciences, Medical University of South Carolina, Charleston, SC, United States.
Department of Medicine, Medical University of South Carolina, Charleston, SC, United States.
Biochem Pharmacol. 2020 Jan;171:113728. doi: 10.1016/j.bcp.2019.113728. Epub 2019 Nov 21.
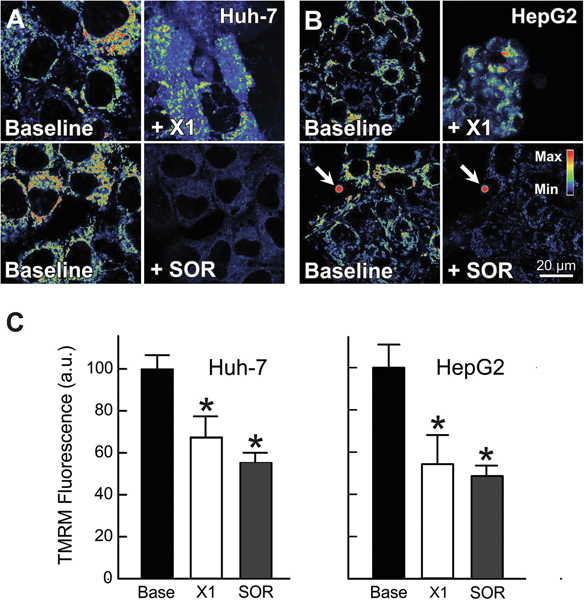
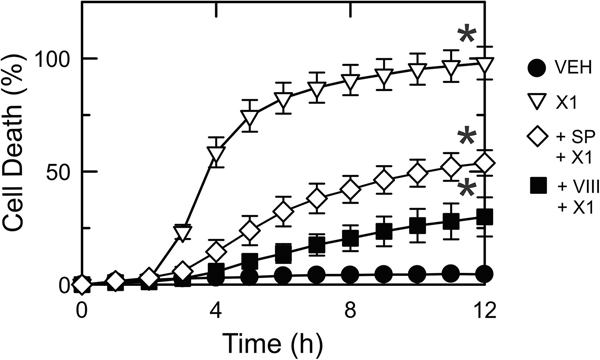
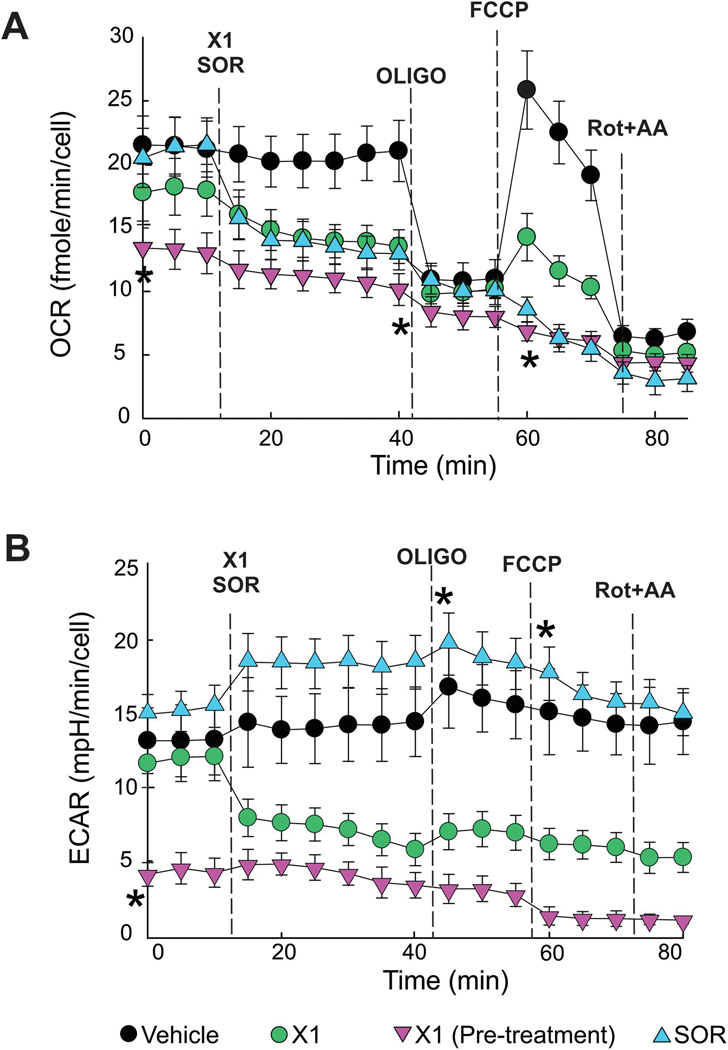
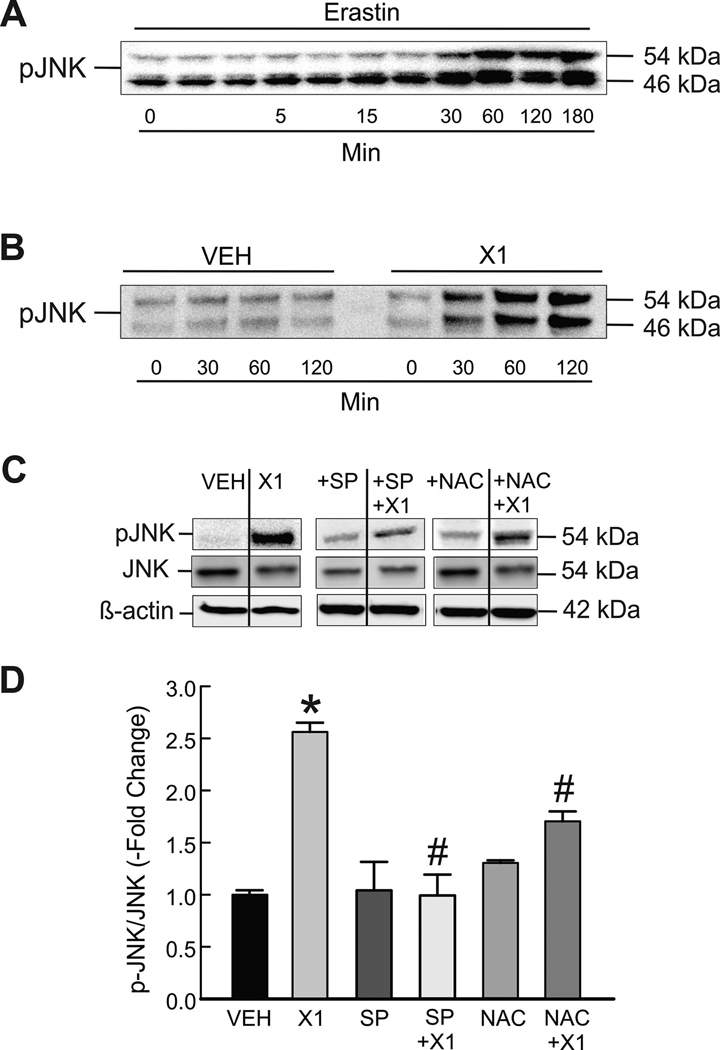
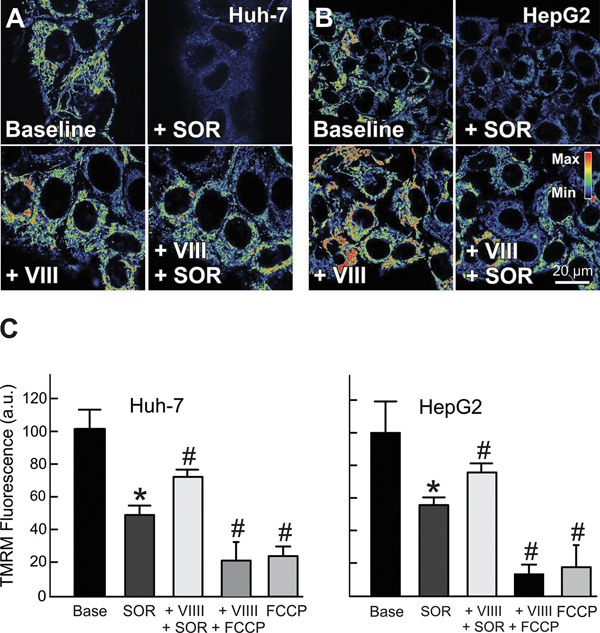
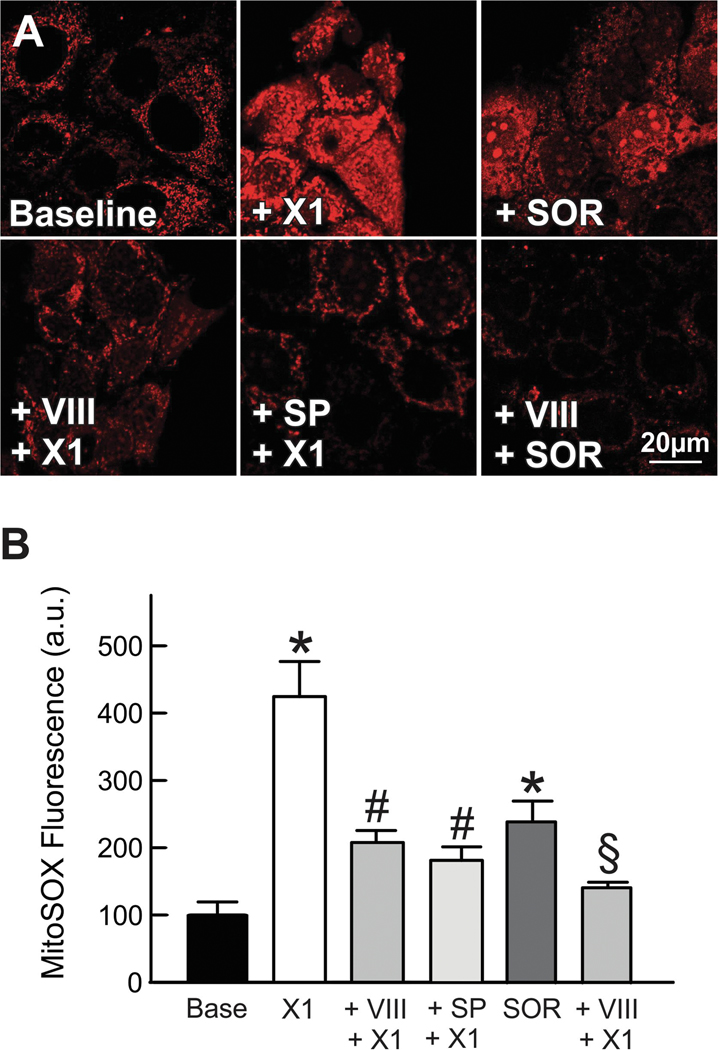
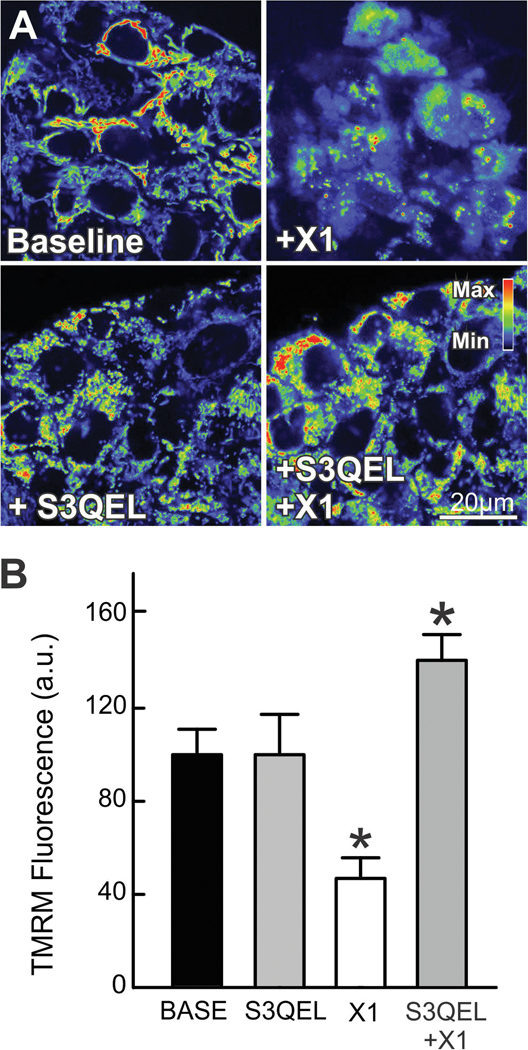
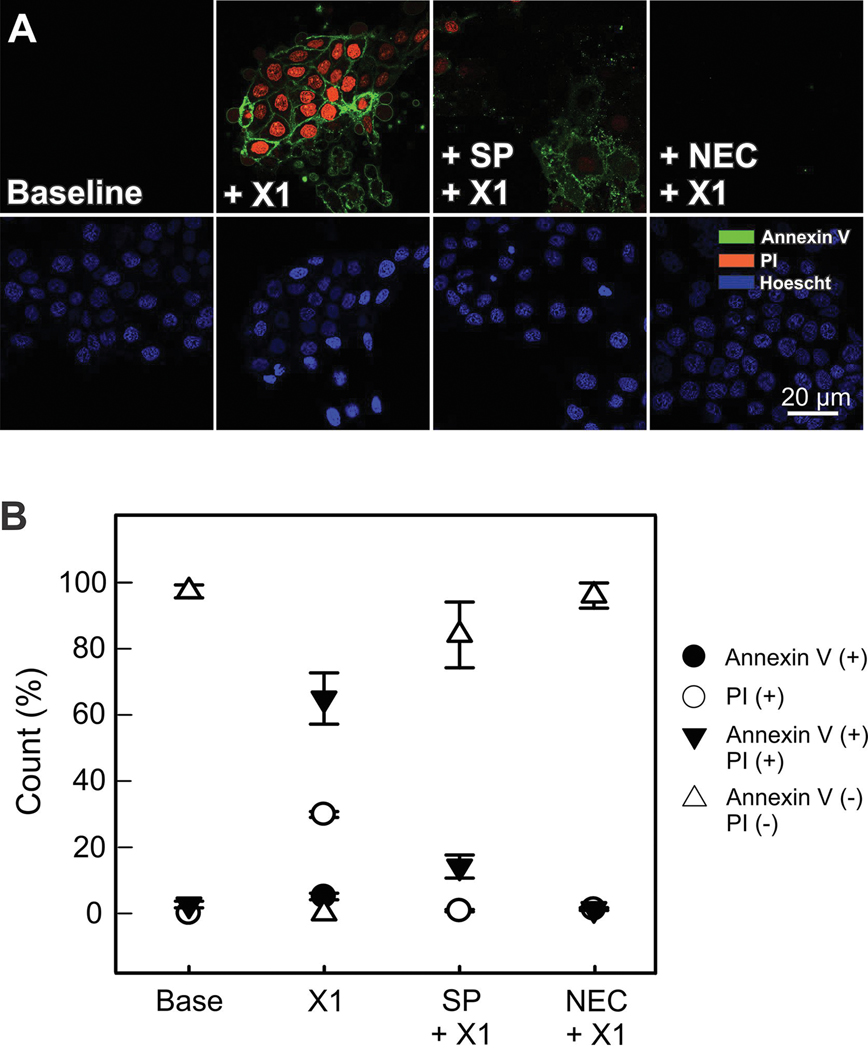
The multikinase inhibitor sorafenib, and opening of voltage dependent anion channels (VDAC) by the erastin-like compound X1 promotes oxidative stress and mitochondrial dysfunction in hepatocarcinoma cells. Here, we hypothesized that X1 and sorafenib induce mitochondrial dysfunction by increasing reactive oxygen species (ROS) formation and activating c-Jun N-terminal kinases (JNKs), leading to translocation of activated JNK to mitochondria. Both X1 and sorafenib increased production of ROS and activated JNK. X1 and sorafenib caused a drop in mitochondrial membrane potential (ΔΨ), a readout of mitochondrial metabolism, after 60 min. Mitochondrial depolarization after X1 and sorafenib occurred in parallel with JNK activation, increased superoxide (O) production, decreased basal and oligomycin sensitive respiration, and decreased maximal respiratory capacity. Increased production of O after X1 or sorafenib was abrogated by JNK inhibition and antioxidants. S3QEL 2, a specific inhibitor of site IIIQ at Complex III, prevented depolarization induced by X1. JNK inhibition by JNK inhibitors VIII and SP600125 also prevented mitochondrial depolarization. After X1, activated JNK translocated to mitochondria as assessed by proximity ligation assays. Tat-Sab KIM1, a peptide selectively preventing the binding of JNK to the outer mitochondrial membrane protein Sab, blocked the depolarization induced by X1 and sorafenib. X1 promoted cell death mostly by necroptosis that was partially prevented by JNK inhibition. These results indicate that JNK activation and translocation to mitochondria is a common mechanism of mitochondrial dysfunction induced by both VDAC opening and sorafenib.
多激酶抑制剂索拉非尼和类似依维莫司的化合物 X1 打开电压依赖性阴离子通道 (VDAC),可促进肝癌细胞的氧化应激和线粒体功能障碍。在这里,我们假设 X1 和索拉非尼通过增加活性氧 (ROS) 的形成和激活 c-Jun N-末端激酶 (JNK) 来诱导线粒体功能障碍,导致激活的 JNK 易位到线粒体。X1 和索拉非尼均增加 ROS 的产生并激活 JNK。X1 和索拉非尼在 60 分钟后引起线粒体膜电位 (ΔΨ) 下降,这是线粒体代谢的读数。X1 和索拉非尼引起的线粒体去极化与 JNK 激活平行发生,增加超氧化物 (O) 的产生,降低基础和寡霉素敏感呼吸,并降低最大呼吸能力。JNK 抑制和抗氧化剂可消除 X1 或索拉非尼后 O 的产生增加。复合物 III 第三 Q 位点的特异性抑制剂 S3QEL 2 可防止 X1 诱导的去极化。JNK 抑制剂 VIII 和 SP600125 抑制 JNK 也可防止线粒体去极化。在用 X1 处理后,通过接近连接测定评估到激活的 JNK 易位到线粒体。Tat-Sab KIM1 是一种选择性阻止 JNK 与外线粒体膜蛋白 Sab 结合的肽,可阻止 X1 和索拉非尼诱导的去极化。X1 诱导的细胞死亡主要通过坏死性凋亡,JNK 抑制可部分阻止该凋亡。这些结果表明,JNK 激活和向线粒体易位是 VDAC 开放和索拉非尼诱导的线粒体功能障碍的共同机制。