Department of Computational Medicine and Bioinformatics, University of Michigan, 100 Washtenaw Avenue, Ann Arbor, MI 48109, USA.
Department of Human Genetics, University of Michigan, 1241 East Catherine Street, Ann Arbor, MI 48109, USA.
Gigascience. 2019 Dec 1;8(12). doi: 10.1093/gigascience/giz153.
Multiple myeloma (MM) is a hematological cancer caused by abnormal accumulation of monoclonal plasma cells in bone marrow. With the increase in treatment options, risk-adapted therapy is becoming more and more important. Survival analysis is commonly applied to study progression or other events of interest and stratify the risk of patients.
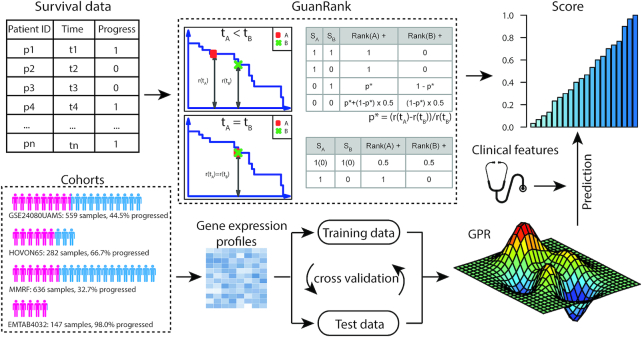
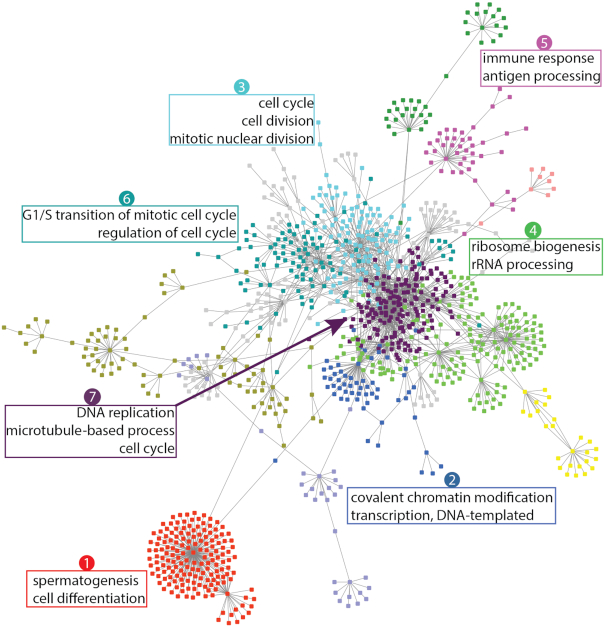
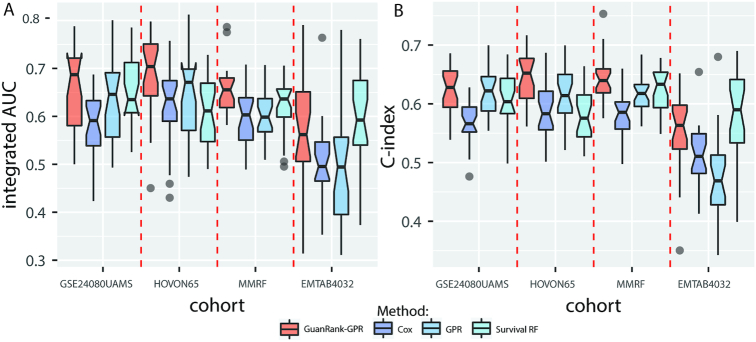
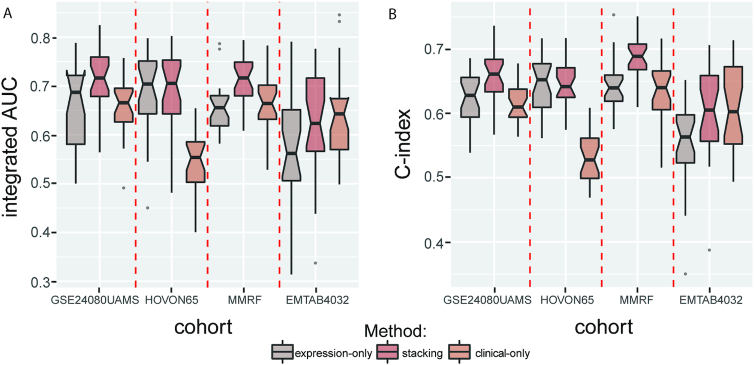
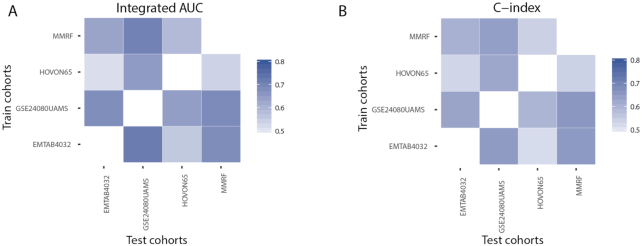
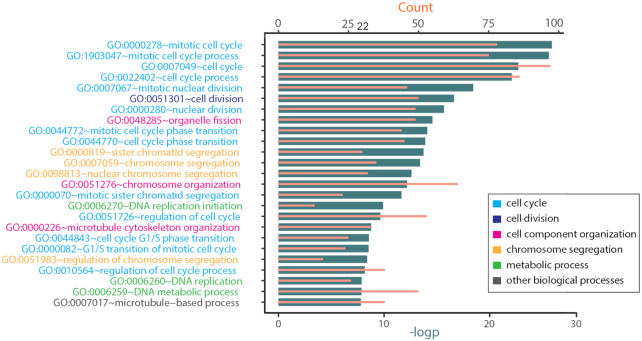
In this study, we present the current state-of-the-art model for MM prognosis and the molecular biomarker set for stratification: the winning algorithm in the 2017 Multiple Myeloma DREAM Challenge, Sub-Challenge 3. Specifically, we built a non-parametric complete hazard ranking model to map the right-censored data into a linear space, where commonplace machine learning techniques, such as Gaussian process regression and random forests, can play their roles. Our model integrated both the gene expression profile and clinical features to predict the progression of MM. Compared with conventional models, such as Cox model and random survival forests, our model achieved higher accuracy in 3 within-cohort predictions. In addition, it showed robust predictive power in cross-cohort validations. Key molecular signatures related to MM progression were identified from our model, which may function as the core determinants of MM progression and provide important guidance for future research and clinical practice. Functional enrichment analysis and mammalian gene-gene interaction network revealed crucial biological processes and pathways involved in MM progression. The model is dockerized and publicly available at https://www.synapse.org/#!Synapse:syn11459638. Both data and reproducible code are included in the docker.
We present the current state-of-the-art prognostic model for MM integrating gene expression and clinical features validated in an independent test set.
多发性骨髓瘤(MM)是一种血液系统癌症,由骨髓中单克隆浆细胞异常积聚引起。随着治疗选择的增加,风险适应治疗变得越来越重要。生存分析通常用于研究进展或其他感兴趣的事件,并对患者的风险进行分层。
在这项研究中,我们提出了 MM 预后的最新模型和分子生物标志物集用于分层:2017 年多发性骨髓瘤 DREAM 挑战赛,第 3 次子挑战赛的获胜算法。具体来说,我们构建了一个非参数完全危险排名模型,将右删失数据映射到线性空间,在该空间中,常见的机器学习技术,如高斯过程回归和随机森林,可以发挥作用。我们的模型整合了基因表达谱和临床特征来预测 MM 的进展。与常规模型(如 Cox 模型和随机生存森林)相比,我们的模型在 3 个同群内预测中具有更高的准确性。此外,它在跨群验证中表现出稳健的预测能力。从我们的模型中鉴定出与 MM 进展相关的关键分子特征,它们可能作为 MM 进展的核心决定因素,并为未来的研究和临床实践提供重要指导。功能富集分析和哺乳动物基因-基因相互作用网络揭示了 MM 进展涉及的关键生物学过程和途径。该模型已被 Docker 化,并可在 https://www.synapse.org/#!Synapse:syn11459638 上公开获取。数据和可重复的代码都包含在 Docker 中。
我们提出了一种将基因表达和临床特征整合在一起的 MM 预后最新模型,并在独立测试集中进行了验证。