Department of Infectious Disease Epidemiology and MRC Centre for Global Infectious Disease Analysis, Imperial College London, Norfolk Place, W2 1PG London, UK.
Department of Mathematical Sciences, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark.
Syst Biol. 2020 Sep 1;69(5):884-896. doi: 10.1093/sysbio/syaa009.
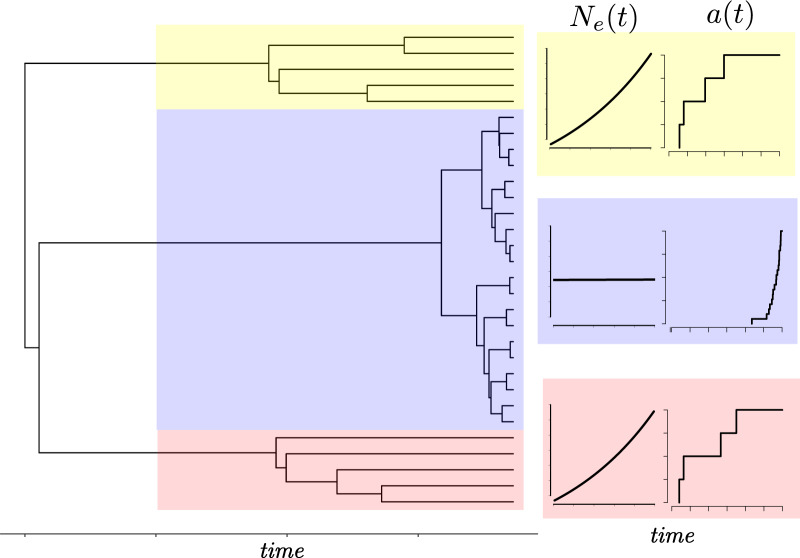
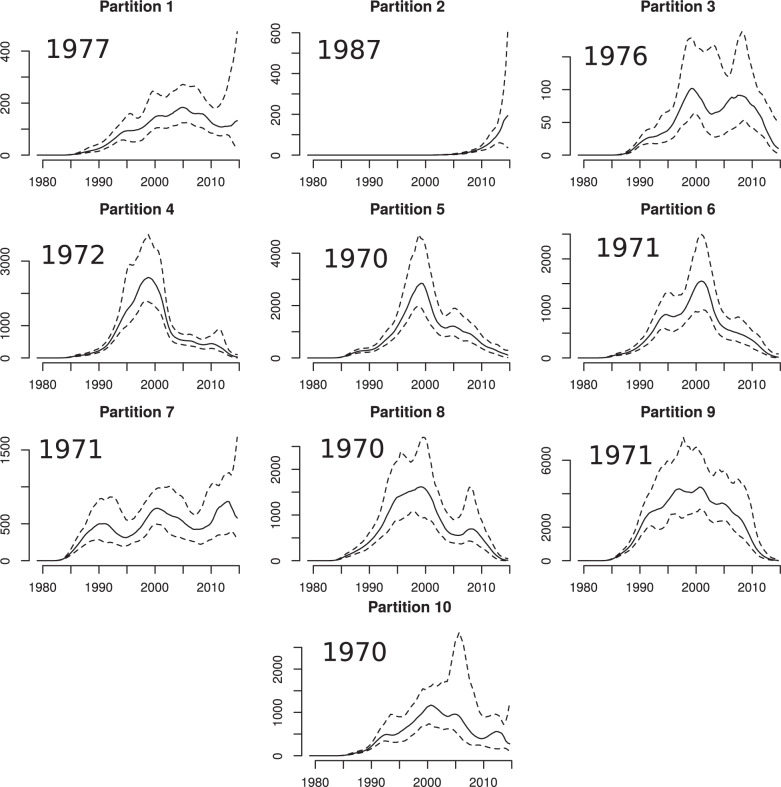
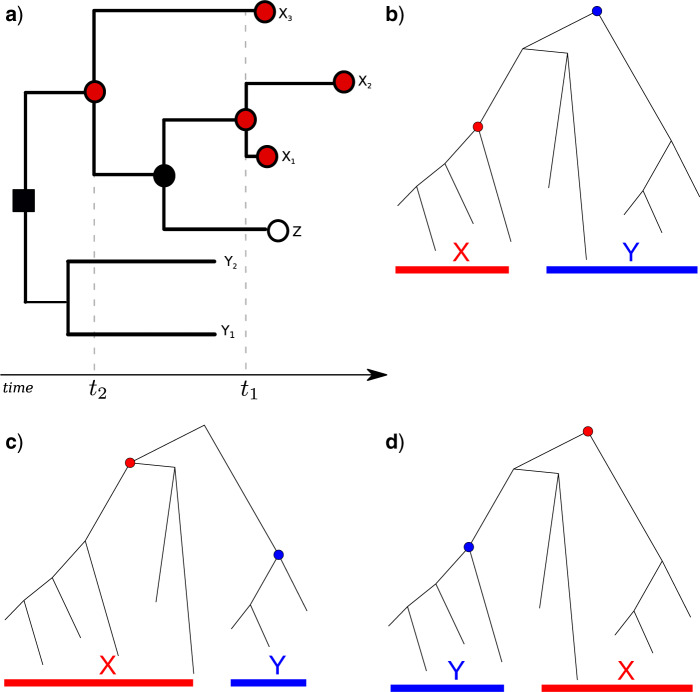
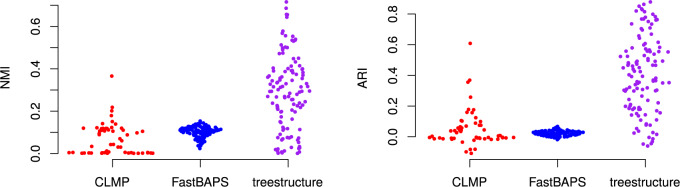
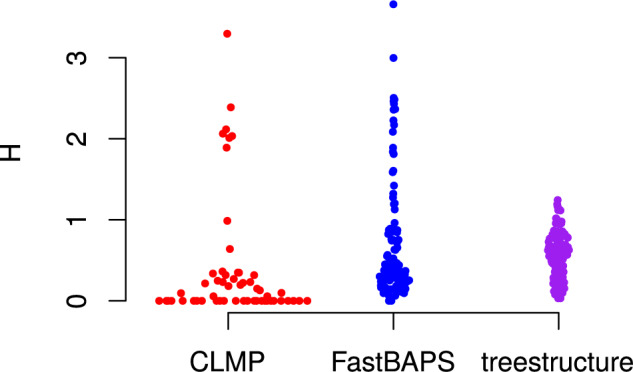
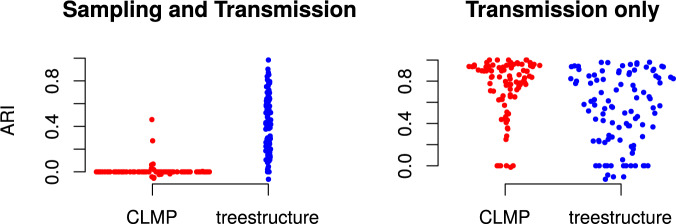
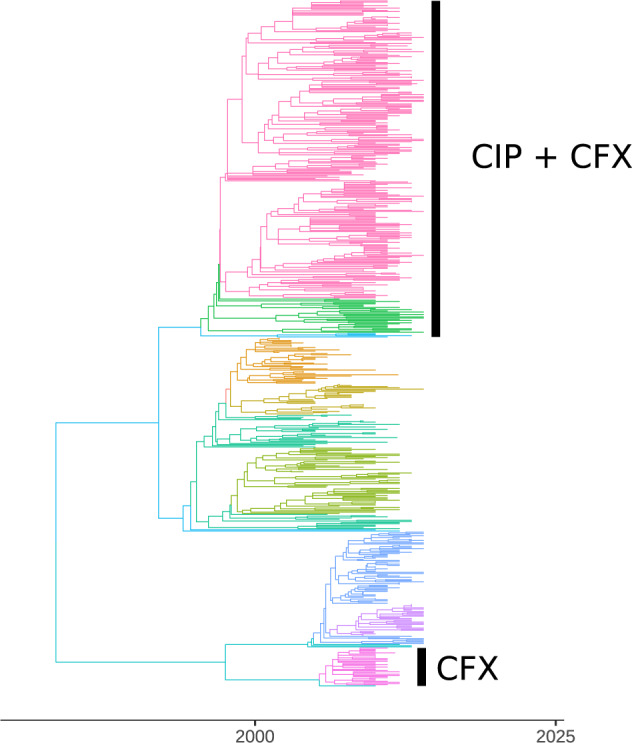
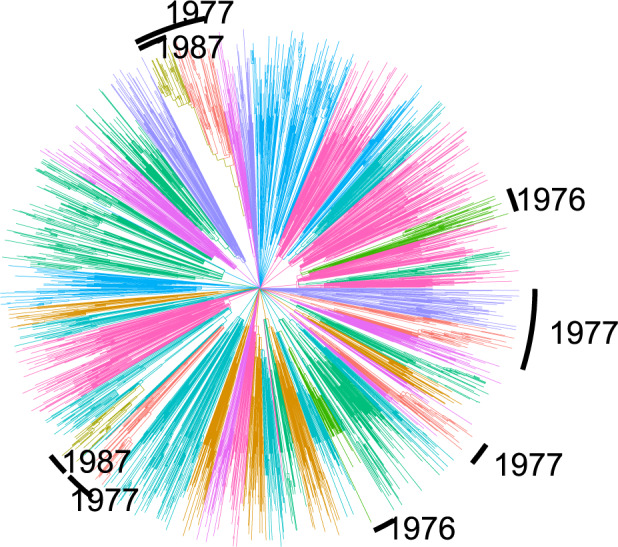
Population structure influences genealogical patterns, however, data pertaining to how populations are structured are often unavailable or not directly observable. Inference of population structure is highly important in molecular epidemiology where pathogen phylogenetics is increasingly used to infer transmission patterns and detect outbreaks. Discrepancies between observed and idealized genealogies, such as those generated by the coalescent process, can be quantified, and where significant differences occur, may reveal the action of natural selection, host population structure, or other demographic and epidemiological heterogeneities. We have developed a fast non-parametric statistical test for detection of cryptic population structure in time-scaled phylogenetic trees. The test is based on contrasting estimated phylogenies with the theoretically expected phylodynamic ordering of common ancestors in two clades within a coalescent framework. These statistical tests have also motivated the development of algorithms which can be used to quickly screen a phylogenetic tree for clades which are likely to share a distinct demographic or epidemiological history. Epidemiological applications include identification of outbreaks in vulnerable host populations or rapid expansion of genotypes with a fitness advantage. To demonstrate the utility of these methods for outbreak detection, we applied the new methods to large phylogenies reconstructed from thousands of HIV-1 partial pol sequences. This revealed the presence of clades which had grown rapidly in the recent past and was significantly concentrated in young men, suggesting recent and rapid transmission in that group. Furthermore, to demonstrate the utility of these methods for the study of antimicrobial resistance, we applied the new methods to a large phylogeny reconstructed from whole genome Neisseria gonorrhoeae sequences. We find that population structure detected using these methods closely overlaps with the appearance and expansion of mutations conferring antimicrobial resistance. [Antimicrobial resistance; coalescent; HIV; population structure.].
人口结构会影响系谱模式,然而,有关人口结构的信息通常是不可用的或无法直接观察到的。在分子流行病学中,推断人口结构非常重要,因为病原体系统发育学越来越多地被用来推断传播模式和检测疫情爆发。观察到的和理想化的系谱之间的差异,例如由合并过程产生的差异,可以进行量化,并且在出现显著差异的情况下,可能揭示自然选择、宿主种群结构或其他人口统计学和流行病学异质性的作用。我们开发了一种快速的非参数统计检验方法,用于检测时间尺度系统发育树上的隐匿种群结构。该检验基于在合并框架内两个分支内共同祖先的理论预期系统发育排序与估计的系统发育之间的对比。这些统计检验还促使开发了可以快速筛选系统发育树以识别可能具有不同人口统计学或流行病学历史的分支的算法。流行病学应用包括识别脆弱宿主群体中的疫情爆发或具有适应优势的基因型的快速扩张。为了证明这些方法在疫情检测中的实用性,我们将新方法应用于从数千个 HIV-1 部分 pol 序列重建的大型系统发育树中。这揭示了在最近过去快速增长的分支的存在,并且在年轻男性中明显集中,表明该群体最近和快速传播。此外,为了证明这些方法在研究抗菌药物耐药性方面的实用性,我们将新方法应用于从全基因组淋病奈瑟氏球菌序列重建的大型系统发育树中。我们发现,这些方法检测到的种群结构与赋予抗菌药物耐药性的突变的出现和扩展密切重叠。[抗菌药物耐药性;合并;HIV;种群结构。]