Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Department of Pathology, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Bioinformatics. 2020 May 1;36(10):3084-3092. doi: 10.1093/bioinformatics/btaa127.
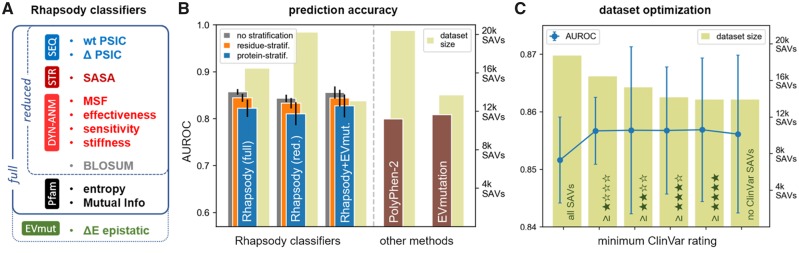
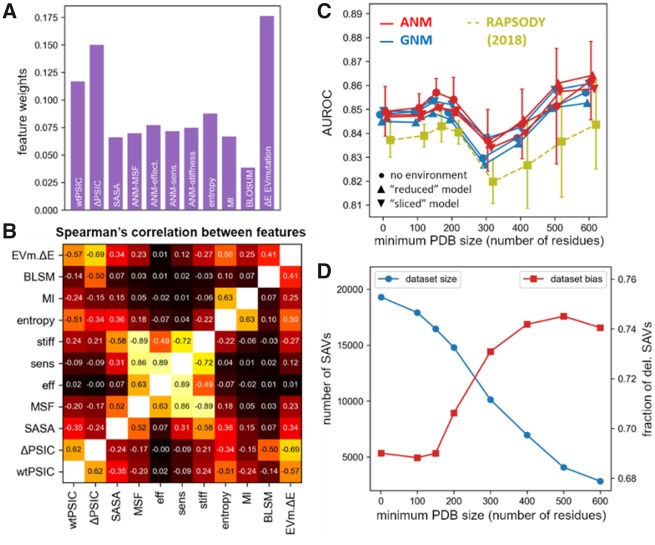
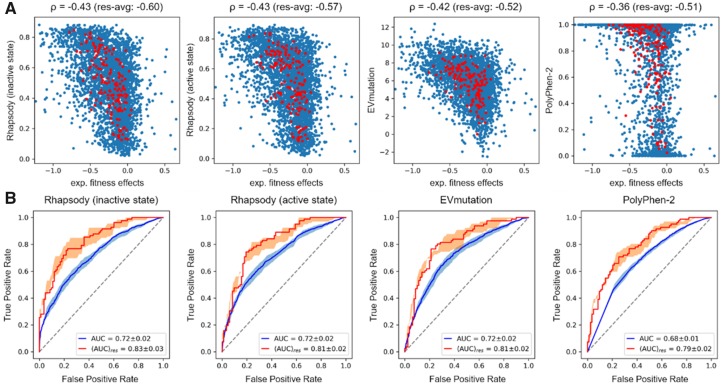
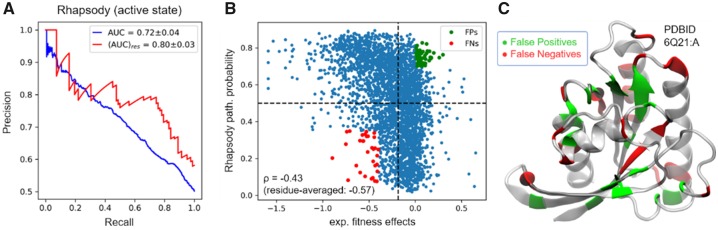
The biological effects of human missense variants have been studied experimentally for decades but predicting their effects in clinical molecular diagnostics remains challenging. Available computational tools are usually based on the analysis of sequence conservation and structural properties of the mutant protein. We recently introduced a new machine learning method that demonstrated for the first time the significance of protein dynamics in determining the pathogenicity of missense variants.
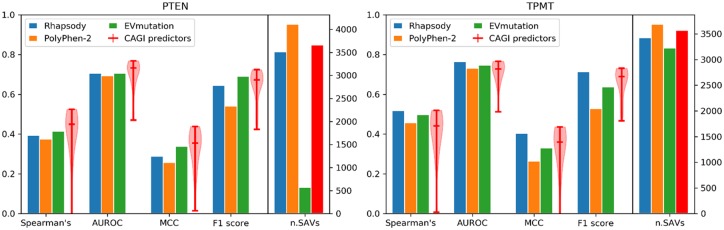
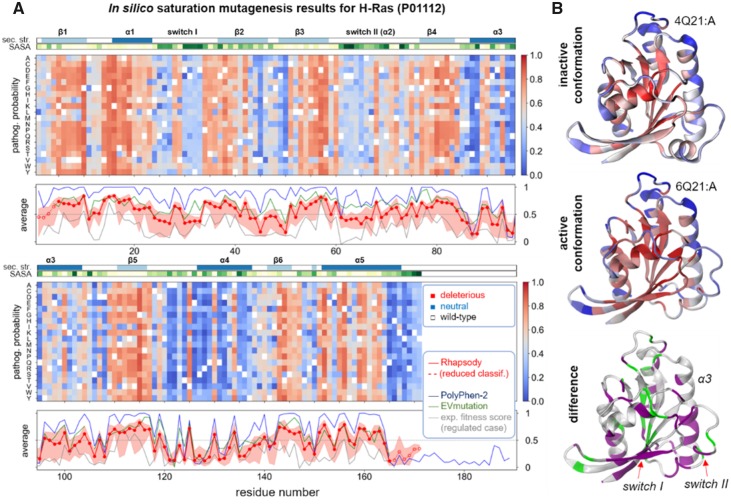
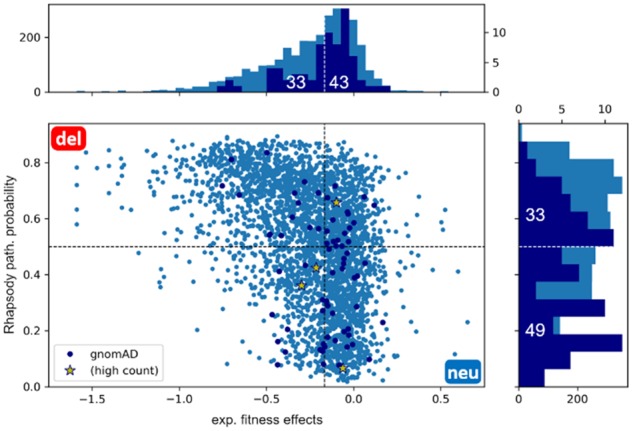
Here, we present a new interface (Rhapsody) that enables fully automated assessment of pathogenicity, incorporating both sequence coevolution data and structure- and dynamics-based features. Benchmarked against a dataset of about 20 000 annotated variants, the methodology is shown to outperform well-established and/or advanced prediction tools. We illustrate the utility of Rhapsody by in silico saturation mutagenesis studies of human H-Ras, phosphatase and tensin homolog and thiopurine S-methyltransferase.
The new tool is available both as an online webserver at http://rhapsody.csb.pitt.edu and as an open-source Python package (GitHub repository: https://github.com/prody/rhapsody; PyPI package installation: pip install prody-rhapsody). Links to additional resources, tutorials and package documentation are provided in the 'Python package' section of the website.
Supplementary data are available at Bioinformatics online.
几十年来,人们一直在对人类错义变异的生物学效应进行实验研究,但预测其在临床分子诊断中的效应仍然具有挑战性。现有的计算工具通常基于对突变蛋白序列保守性和结构特性的分析。我们最近引入了一种新的机器学习方法,该方法首次证明了蛋白质动力学在确定错义变异的致病性方面的重要性。
在这里,我们提出了一个新的界面(Rhapsody),它能够自动评估致病性,同时结合序列共进化数据和基于结构和动力学的特征。该方法在大约 20000 个注释变异的数据集上进行了基准测试,结果表明其性能优于成熟的和/或先进的预测工具。我们通过对人类 H-Ras、磷酸酶和张力蛋白同系物和巯基嘌呤 S-甲基转移酶的体外饱和诱变研究说明了 Rhapsody 的实用性。
新工具既可以作为在线网络服务器(http://rhapsody.csb.pitt.edu)使用,也可以作为开源 Python 包(GitHub 存储库:https://github.com/prody/rhapsody;PyPI 包安装:pip install prody-rhapsody)使用。在网站的“Python 包”部分提供了其他资源、教程和包文档的链接。
补充数据可在 Bioinformatics 在线获得。