Firrincieli Andrea, Khorasani Mahsa, Frank A Carolin, Doty Sharon Lafferty
School of Environmental and Forest Sciences, College of the Environment, University of Washington, Seattle, WA, United States.
Life & Environmental Sciences School of Natural Sciences, University of California, Merced, Merced, CA, United States.
Front Plant Sci. 2020 Feb 28;11:203. doi: 10.3389/fpls.2020.00203. eCollection 2020.
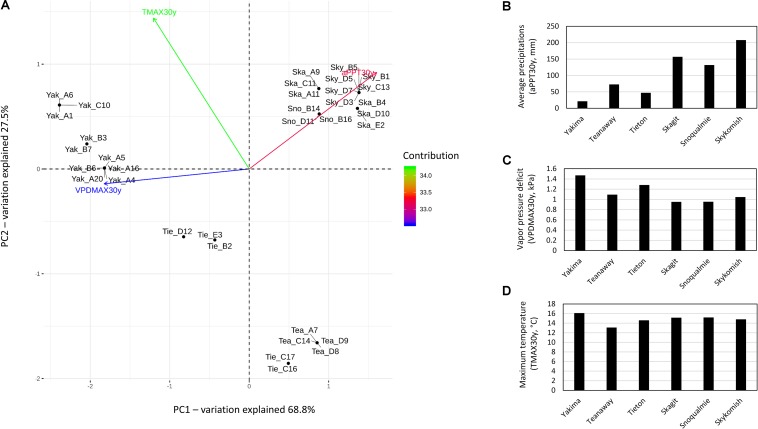
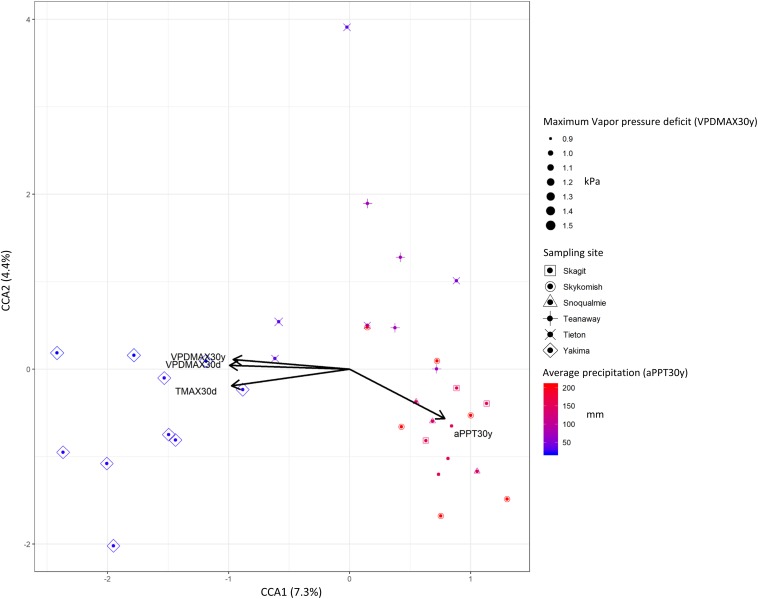
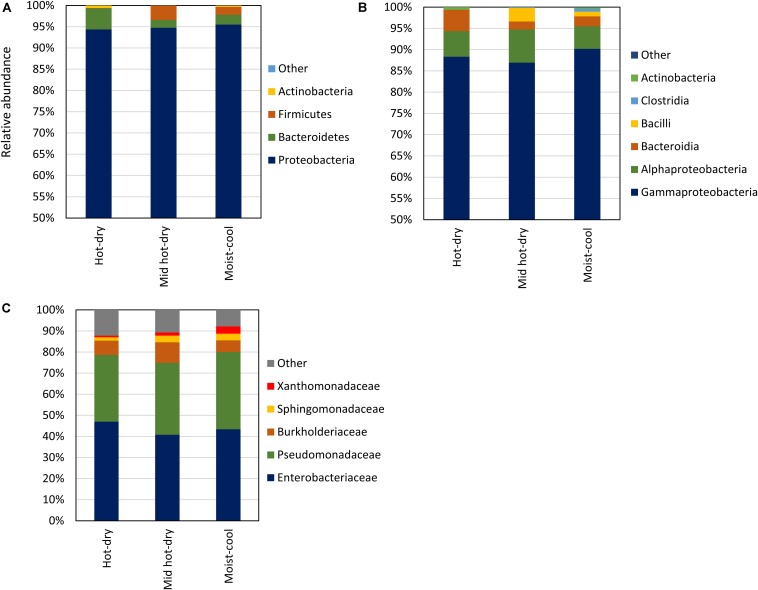
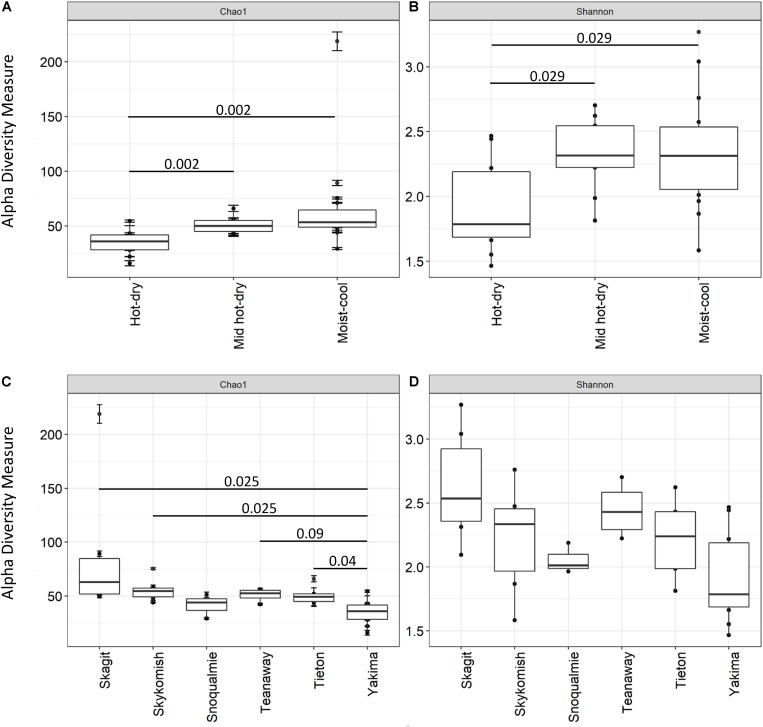
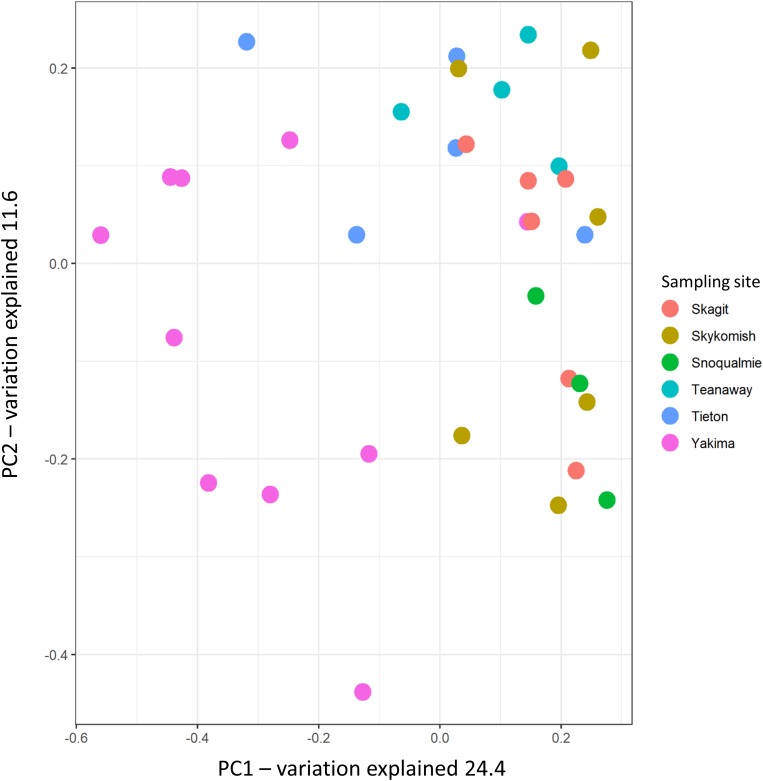
Plant-associated microbial communities play a central role in the plant response to biotic and abiotic stimuli, improving plant fitness under challenging growing conditions. Many studies have focused on the characterization of changes in abundance and composition of root-associated microbial communities as a consequence of the plant response to abiotic factors such as altered soil nutrients and drought. However, changes in composition in response to abiotic factors are still poorly understood concerning the endophytic community associated to the phyllosphere, the above-ground plant tissues. In the present study, we applied high-throughput 16S rDNA gene sequencing of the phyllosphere endophytic bacterial communities colonizing wild (black cottonwood) plants growing in native, nutrient-limited environments characterized by hot-dry (xeric) riparian zones (Yakima River, WA), riparian zones with mid hot-dry (Tieton and Teanaway Rivers, WA) and moist (mesic) climates (Snoqualmie, Skykomish and Skagit Rivers, WA). From sequencing data, 587 Amplicon Sequence Variants (ASV) were identified. Surprisingly, our data show that a core microbiome could be found in phyllosphere-associated endophytic communities in trees growing on opposite sides of the Cascades Mountain Range. Considering only taxa appearing in at least 90% of all samples within each climatic zone, the core microbiome was dominated only by two ASVs affiliated and two ASVs of the family. Alpha-diversity measures indicated that plants colonizing hot-dry environments showed a lower diversity than those from mid hot-dry and moist climates. Beta-diversity measures showed that bacterial composition was significantly different across sampling sites. Accordingly, we found that specific ASV affiliated to and were significantly more abundant in the phyllosphere endophytic community colonizing plants adapted to the xeric environment. In summary, this study highlights that sampling site is the major driver of variation and that only a few ASV showed a distribution that significantly correlated to climate variables.
与植物相关的微生物群落对植物应对生物和非生物刺激起着核心作用,在具有挑战性的生长条件下提高植物的适应性。许多研究聚焦于植物对非生物因素(如土壤养分变化和干旱)作出反应时,根际相关微生物群落丰度和组成的变化特征。然而,关于叶际(地上植物组织)相关内生群落对非生物因素的组成变化,我们仍知之甚少。在本研究中,我们对生长在以炎热干燥(旱生)河岸带(华盛顿州亚基马河)、中等炎热干燥(华盛顿州蒂顿河和蒂纳韦河)和湿润(中生)气候(华盛顿州斯诺夸尔米河、斯基科米什河和斯卡吉特河)为特征的原生、养分有限环境中的野生(黑杨)植物叶际内生细菌群落进行了高通量16S rDNA基因测序。从测序数据中,鉴定出587个扩增子序列变体(ASV)。令人惊讶的是,我们的数据表明,在喀斯喀特山脉两侧生长的树木的叶际相关内生群落中可以发现一个核心微生物组。仅考虑在每个气候区内至少90%的样本中出现的分类群,核心微生物组仅由两个属于 的ASV和 科的两个ASV主导。α多样性测量表明,生长在炎热干燥环境中的植物多样性低于中等炎热干燥和湿润气候中的植物。β多样性测量表明,不同采样地点的细菌组成存在显著差异。因此,我们发现,属于 和 的特定ASV在适应旱生环境的植物叶际内生群落中显著更为丰富。总之,本研究强调采样地点是变异的主要驱动因素,只有少数ASV的分布与气候变量显著相关。