Khoury College of Computer Sciences, Northeastern University, Boston, Massachusetts.
Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center Basel, Basel, Switzerland.
Mol Cell Proteomics. 2020 Jun;19(6):944-959. doi: 10.1074/mcp.RA119.001792. Epub 2020 Mar 31.
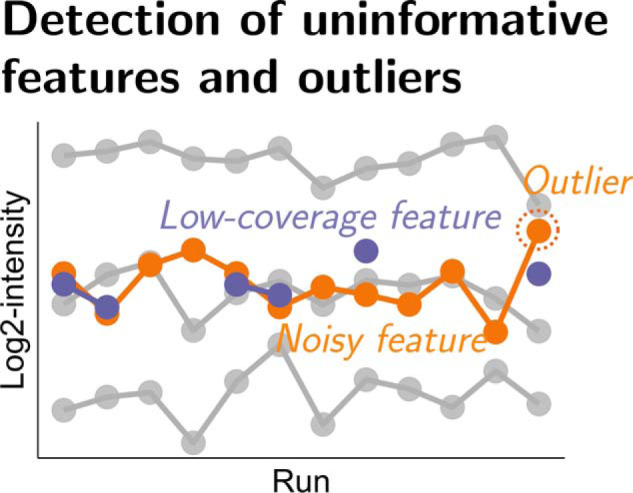
In bottom-up mass spectrometry-based proteomics, relative protein quantification is often achieved with data-dependent acquisition (DDA), data-independent acquisition (DIA), or selected reaction monitoring (SRM). These workflows quantify proteins by summarizing the abundances of all the spectral features of the protein ( precursor ions, transitions or fragments) in a single value per protein per run. When abundances of some features are inconsistent with the overall protein profile (for technological reasons such as interferences, or for biological reasons such as post-translational modifications), the protein-level summaries and the downstream conclusions are undermined. We propose a statistical approach that automatically detects spectral features with such inconsistent patterns. The detected features can be separately investigated, and if necessary, removed from the data set. We evaluated the proposed approach on a series of benchmark-controlled mixtures and biological investigations with DDA, DIA and SRM data acquisitions. The results demonstrated that it could facilitate and complement manual curation of the data. Moreover, it can improve the estimation accuracy, sensitivity and specificity of detecting differentially abundant proteins, and reproducibility of conclusions across different data processing tools. The approach is implemented as an option in the open-source R-based software MSstats.
在基于自上而下的质谱的蛋白质组学中,相对蛋白质定量通常通过数据依赖采集(DDA)、数据独立采集(DIA)或选择反应监测(SRM)来实现。这些工作流程通过对每个蛋白质在每个运行中所有蛋白质谱特征(前体离子、转换或片段)的丰度进行总结来定量蛋白质。当某些特征的丰度与整体蛋白质谱不一致时(由于技术原因,如干扰,或由于生物原因,如翻译后修饰),蛋白质水平的总结和下游结论就会受到影响。我们提出了一种统计方法,该方法可以自动检测具有这种不一致模式的谱特征。可以分别研究检测到的特征,如果有必要,可以从数据集中删除。我们在一系列基于 DDA、DIA 和 SRM 数据采集的基准对照混合物和生物学研究中评估了所提出的方法。结果表明,它可以促进和补充数据的手动整理。此外,它可以提高检测差异丰度蛋白质的估计准确性、灵敏度和特异性,以及不同数据处理工具之间结论的可重复性。该方法作为开源基于 R 的软件 MSstats 的一个选项实现。