Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 11724, USA.
Nat Commun. 2020 Apr 14;11(1):1782. doi: 10.1038/s41467-020-15512-5.
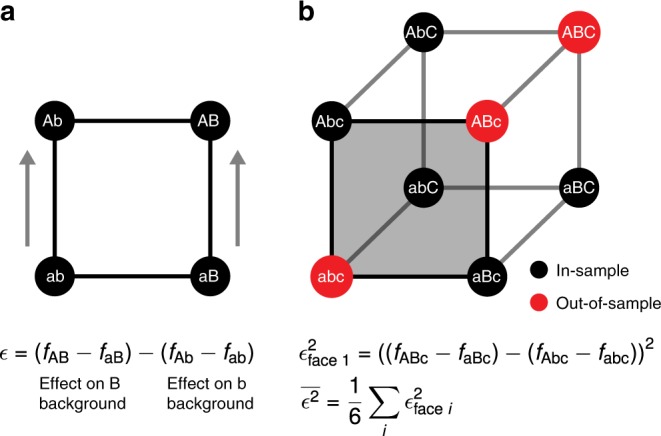
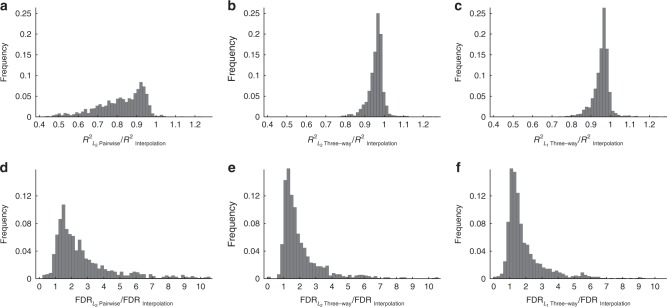
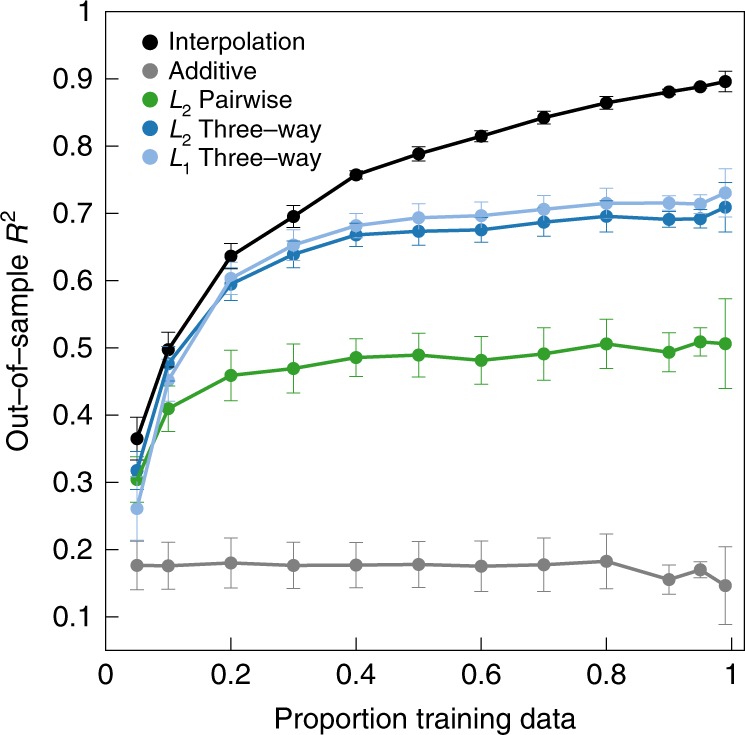
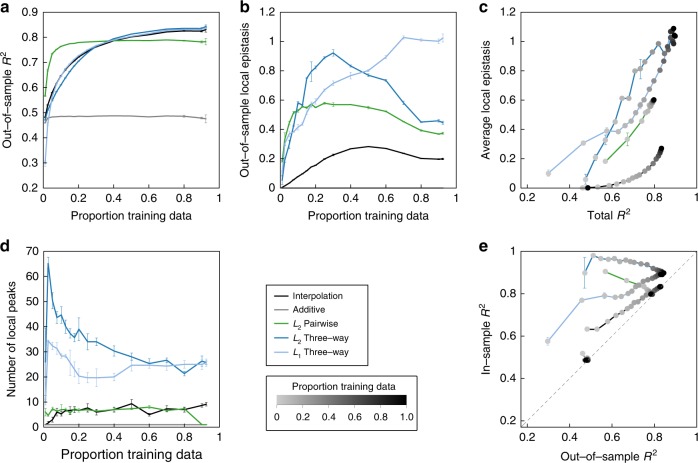
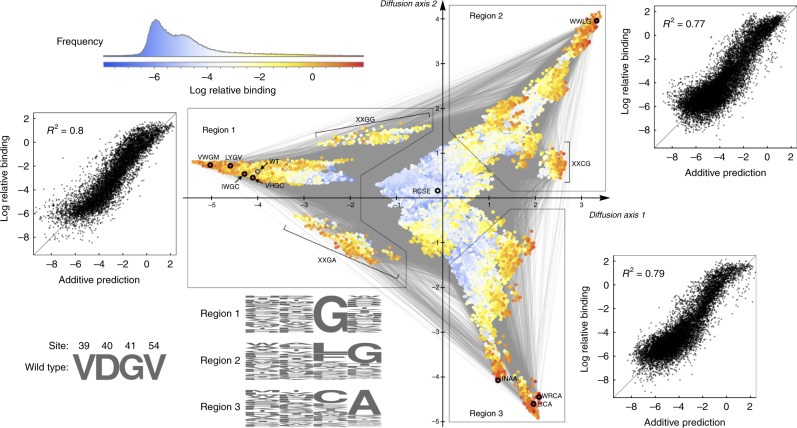
Massively parallel phenotyping assays have provided unprecedented insight into how multiple mutations combine to determine biological function. While such assays can measure phenotypes for thousands to millions of genotypes in a single experiment, in practice these measurements are not exhaustive, so that there is a need for techniques to impute values for genotypes whose phenotypes have not been directly assayed. Here, we present an imputation method based on inferring the least epistatic possible sequence-function relationship compatible with the data. In particular, we infer the reconstruction where mutational effects change as little as possible across adjacent genetic backgrounds. The resulting models can capture complex higher-order genetic interactions near the data, but approach additivity where data is sparse or absent. We apply the method to high-throughput transcription factor binding assays and use it to explore a fitness landscape for protein G.
大规模并行表型分析为研究多种突变如何组合来决定生物功能提供了前所未有的见解。虽然此类分析可以在单次实验中测量数千到数百万种基因型的表型,但实际上这些测量并非详尽无遗,因此需要有技术来推断那些表型尚未直接测定的基因型的值。在这里,我们提出了一种基于推断与数据兼容的最小上位性可能的序列-功能关系的推断方法。具体来说,我们推断出突变效应在相邻遗传背景下变化最小的重建。由此产生的模型可以捕捉到数据附近复杂的高阶遗传相互作用,但在数据稀疏或不存在的情况下接近加性。我们将该方法应用于高通量转录因子结合分析,并使用它来探索蛋白质 G 的适应度景观。