Martínez-Álvaro Marina, Auffret Marc D, Stewart Robert D, Dewhurst Richard J, Duthie Carol-Anne, Rooke John A, Wallace R John, Shih Barbara, Freeman Tom C, Watson Mick, Roehe Rainer
Scotland's Rural College, Edinburgh, United Kingdom.
Institute for Animal Science and Technology, Polytechnic University of Valencia, Valencia, Spain.
Front Microbiol. 2020 Apr 17;11:659. doi: 10.3389/fmicb.2020.00659. eCollection 2020.
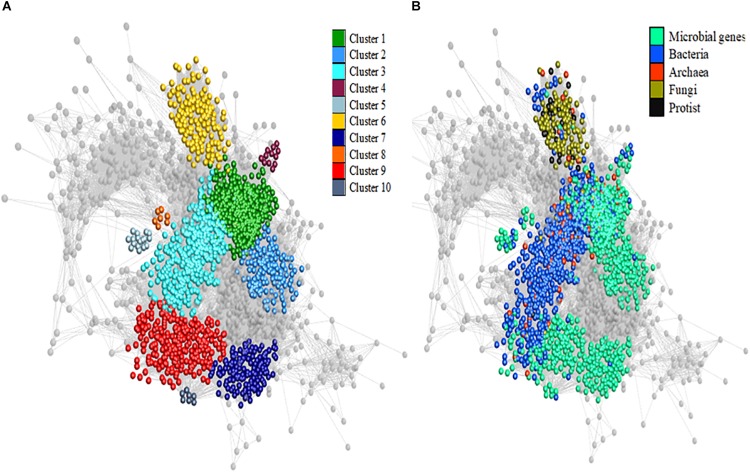
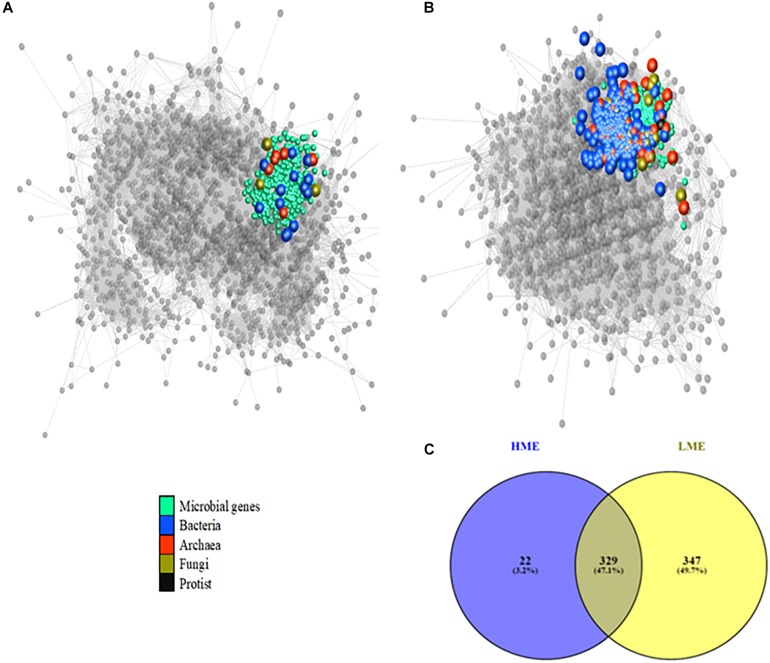
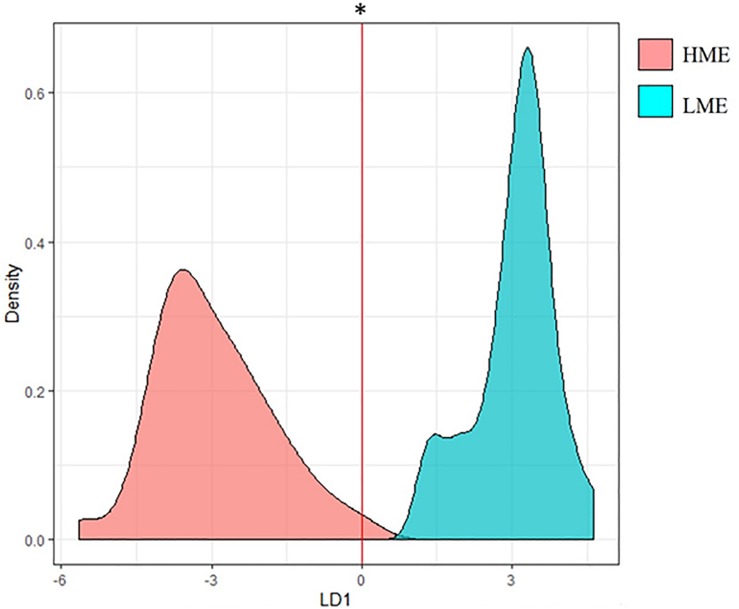
A network analysis including relative abundances of all ruminal microbial genera (archaea, bacteria, fungi, and protists) and their genes was performed to improve our understanding of how the interactions within the ruminal microbiome affects methane emissions (CH). Metagenomics and CH data were available from 63 bovines of a two-breed rotational cross, offered two basal diets. Co-abundance network analysis revealed 10 clusters of functional niches. The most abundant hydrogenotrophic with key microbial genes involved in methanogenesis occupied a different functional niche (i.e., "methanogenesis" cluster) than methylotrophic (Candidatus ) and acetogens (). Fungi and protists clustered together and other plant fiber degraders like occupied a seperate cluster. A Partial Least Squares analysis approach to predict CH variation in each cluster showed the methanogenesis cluster had the best prediction ability (57.3%). However, the most important explanatory variables in this cluster were genes involved in complex carbohydrate degradation, metabolism of sugars and amino acids and Candidatus carrying nitrogen fixation genes, but not methanogenic archaea and their genes. The cluster containing , isolated from other microorganisms, was positively associated with CH and explained 49.8% of its variability, showing fermentative advantages compared to other bacteria and fungi in providing substrates (e.g., formate) for methanogenesis. In other clusters, genes with enhancing effect on CH were related to lactate and butyrate ( and ) production and simple amino acids metabolism. In comparison, ruminal genes negatively related to CH were involved in carbohydrate degradation via lactate and succinate and synthesis of more complex amino acids by γ-Proteobacteria. When analyzing low- and high-methane emitters data in separate networks, competition between methanogens in the methanogenesis cluster was uncovered by a broader diversity of methanogens involved in the three methanogenesis pathways and larger interactions within and between communities in low compared to high emitters. Generally, our results suggest that differences in CH are mainly explained by other microbial communities and their activities rather than being only methanogens-driven. Our study provides insight into the interactions of the rumen microbial communities and their genes by uncovering functional niches affecting CH, which will benefit the development of efficient CH mitigation strategies.
进行了一项网络分析,包括所有瘤胃微生物属(古菌、细菌、真菌和原生生物)及其基因的相对丰度,以增进我们对瘤胃微生物组内的相互作用如何影响甲烷排放(CH)的理解。宏基因组学和CH数据来自一个两品种轮回杂交的63头奶牛,它们被投喂两种基础日粮。共丰度网络分析揭示了10个功能生态位簇。与产甲烷作用相关的最丰富的氢营养型以及关键微生物基因,与甲基营养型(“Ca. Methylomirabilis oxyfera”)和产乙酸菌(“Acetobacterium”)占据不同的功能生态位(即“产甲烷作用”簇)。真菌和原生生物聚集在一起,而其他植物纤维降解菌如“Ruminococcus”占据一个单独的簇。一种用于预测每个簇中CH变化的偏最小二乘分析方法表明,产甲烷作用簇具有最佳的预测能力(57.3%)。然而,该簇中最重要的解释变量是参与复杂碳水化合物降解、糖和氨基酸代谢的基因以及携带固氮基因的“Ca. Methylomirabilis oxyfera”,而非产甲烷古菌及其基因。与其他微生物分离的包含“Succiniclasticum”的簇与CH呈正相关,并解释了其49.8%的变异性,表明在为产甲烷作用提供底物(如甲酸盐)方面,与其他细菌和真菌相比具有发酵优势。在其他簇中,对CH有增强作用的基因与乳酸和丁酸(“Megasphaera”和“Succiniclasticum”)的产生以及简单氨基酸代谢有关。相比之下,与CH呈负相关的瘤胃基因参与通过乳酸和琥珀酸进行的碳水化合物降解以及γ-变形菌合成更复杂的氨基酸。在单独的网络中分析低甲烷排放者和高甲烷排放者的数据时,发现产甲烷作用簇中产甲烷菌之间的竞争,低排放者中参与三种产甲烷途径的产甲烷菌种类更多,且群落内部和群落之间的相互作用比高排放者更大。总体而言,我们的结果表明,CH的差异主要由其他微生物群落及其活动解释,而非仅由产甲烷菌驱动。我们的研究通过揭示影响CH的功能生态位,深入了解了瘤胃微生物群落及其基因之间的相互作用,这将有利于高效CH减排策略的发展。