Section of Neurobiology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Division of Pediatric Emergency, Department of Pediatrics, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Int J Mol Sci. 2020 May 7;21(9):3297. doi: 10.3390/ijms21093297.
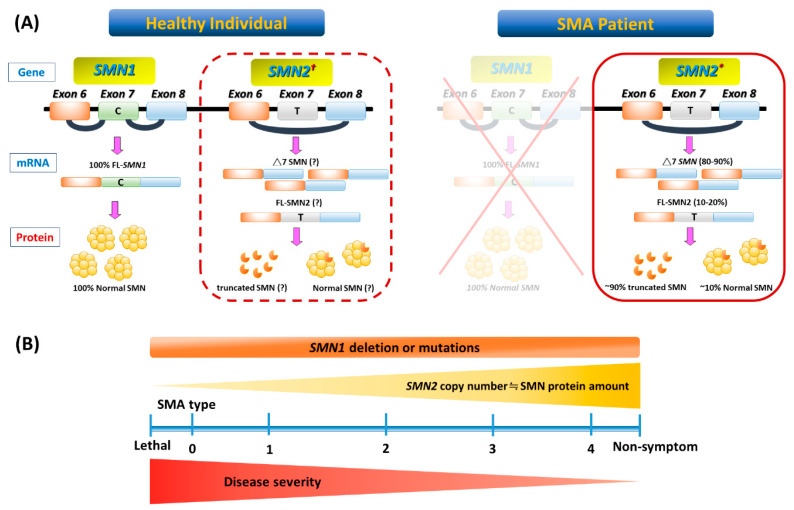
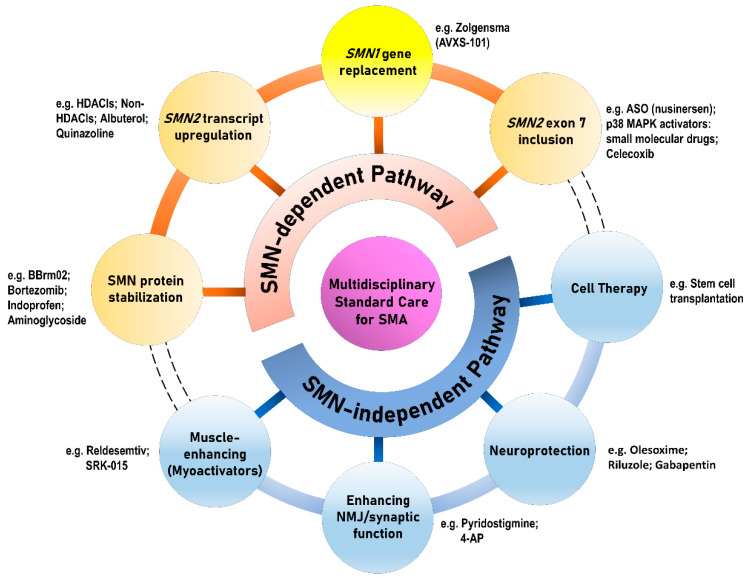
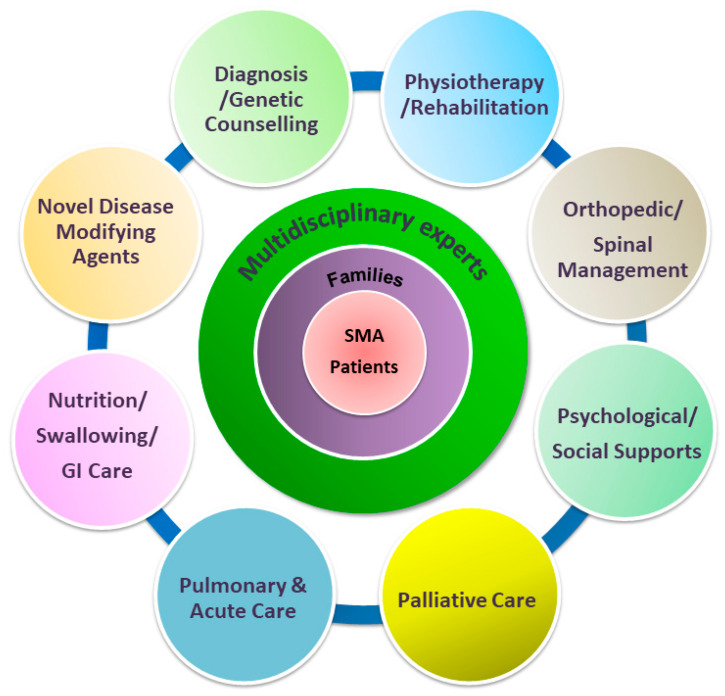
Spinal muscular atrophy (SMA) is a congenital neuromuscular disorder characterized by motor neuron loss, resulting in progressive weakness. SMA is notable in the health care community because it accounts for the most common cause of infant death resulting from a genetic defect. SMA is caused by low levels of the survival motor neuron protein (SMN) resulting from gene mutations or deletions. However, patients always harbor various copies of , an almost identical but functionally deficient copy of the gene. A genotype-phenotype correlation suggests that is a potent disease modifier for SMA, which also represents the primary target for potential therapies. Increasing comprehension of SMA pathophysiology, including the characterization of and genes and SMN protein functions, has led to the development of multiple therapeutic approaches. Until the end of 2016, no cure was available for SMA, and management consisted of supportive measures. Two breakthrough SMN-targeted treatments, either using antisense oligonucleotides (ASOs) or virus-mediated gene therapy, have recently been approved. These two novel therapeutics have a common objective: to increase the production of SMN protein in MNs and thereby improve motor function and survival. However, neither therapy currently provides a complete cure. Treating patients with SMA brings new responsibilities and unique dilemmas. As SMA is such a devastating disease, it is reasonable to assume that a unique therapeutic solution may not be sufficient. Current approaches under clinical investigation differ in administration routes, frequency of dosing, intrathecal versus systemic delivery, and mechanisms of action. Besides, emerging clinical trials evaluating the efficacy of either SMN-dependent or SMN-independent approaches are ongoing. This review aims to address the different knowledge gaps between genotype, phenotypes, and potential therapeutics.
脊髓性肌萎缩症(SMA)是一种先天性神经肌肉疾病,其特征是运动神经元丢失,导致进行性肌无力。在医疗保健界,SMA 很重要,因为它是导致由遗传缺陷引起的婴儿死亡的最常见原因。SMA 是由于生存运动神经元蛋白(SMN)水平降低引起的,这是由于基因突变或缺失。然而,患者总是携带各种 的副本, 是基因的几乎相同但功能缺陷的副本。基因型-表型相关性表明, 是 SMA 的一种有效疾病修饰物,也是潜在治疗方法的主要靶点。对 SMA 病理生理学的理解不断增加,包括 和 基因以及 SMN 蛋白功能的特征,导致了多种治疗方法的发展。直到 2016 年底,SMA 还没有治愈方法,治疗方法包括支持性措施。最近,两种突破性的 SMN 靶向治疗方法,一种是使用反义寡核苷酸(ASO),另一种是病毒介导的基因治疗,已经获得批准。这两种新的治疗方法有一个共同的目标:增加 MN 中 SMN 蛋白的产生,从而改善运动功能和存活。然而,目前没有一种治疗方法能完全治愈 SMA。治疗 SMA 患者带来了新的责任和独特的困境。由于 SMA 是一种如此毁灭性的疾病,因此可以合理地假设,独特的治疗方法可能是不够的。目前正在临床研究中的方法在给药途径、给药频率、鞘内与全身给药以及作用机制上有所不同。此外,正在进行评估 SMN 依赖或非 SMN 依赖方法疗效的新兴临床试验。本综述旨在解决基因型、表型和潜在治疗方法之间的不同知识差距。