Department of Molecular and Cellular Physiology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Schrödinger, New York, NY 10036, USA.
Structure. 2020 Jun 2;28(6):707-716.e3. doi: 10.1016/j.str.2020.04.018. Epub 2020 May 14.
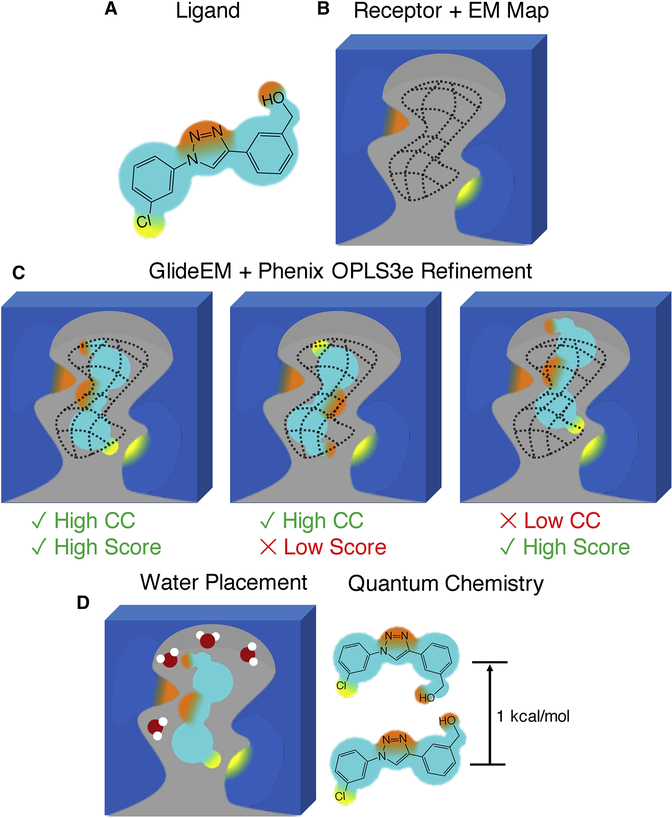
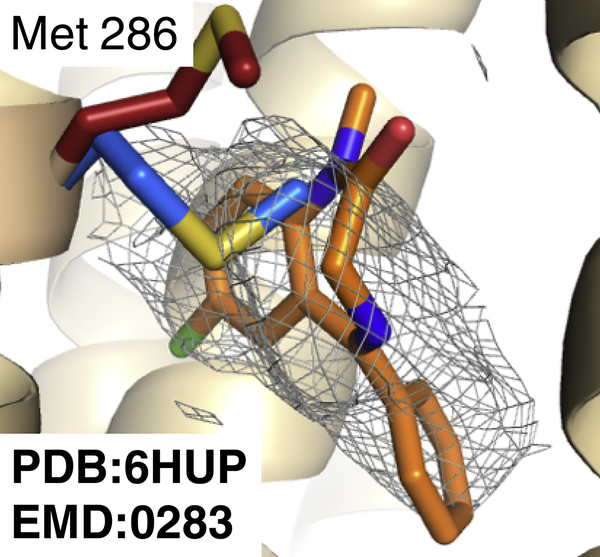
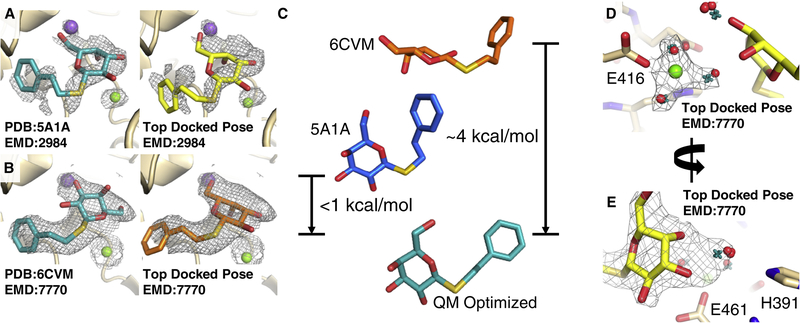
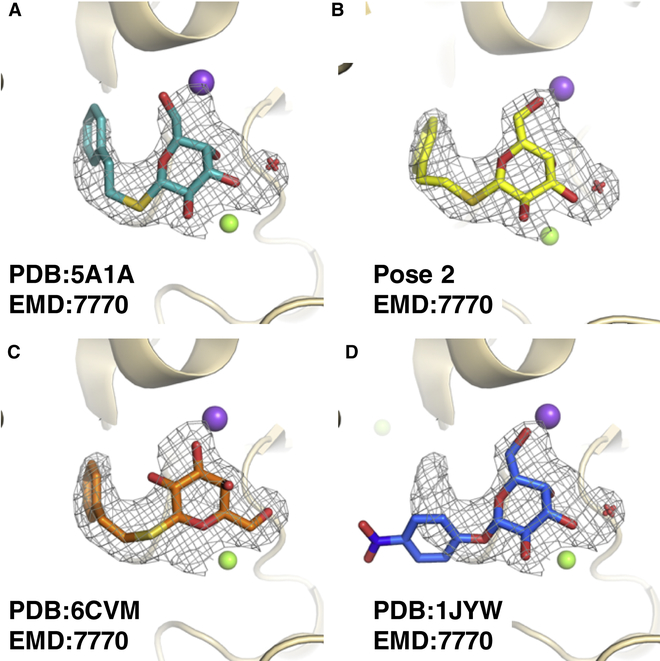
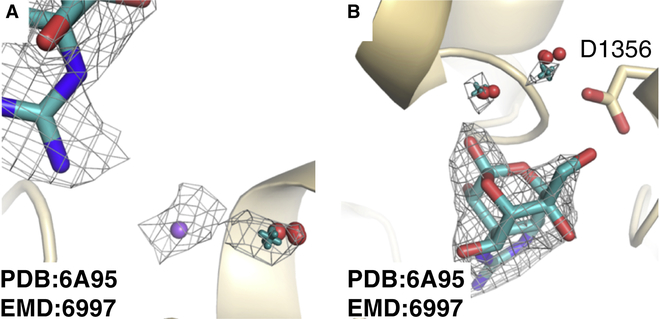
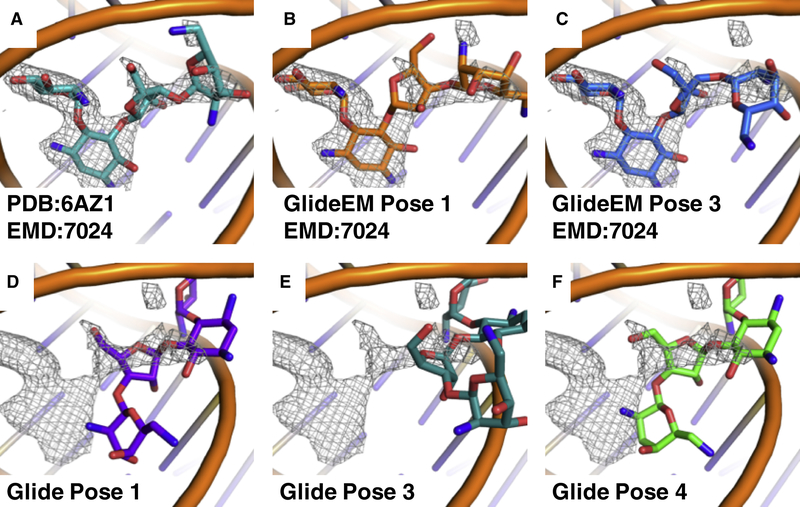
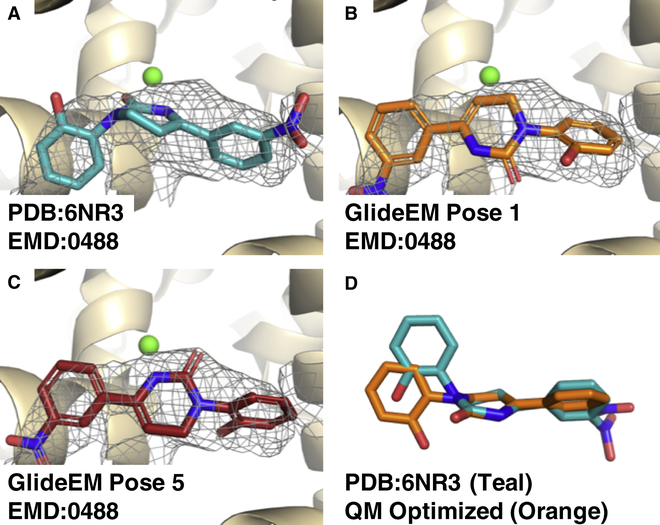
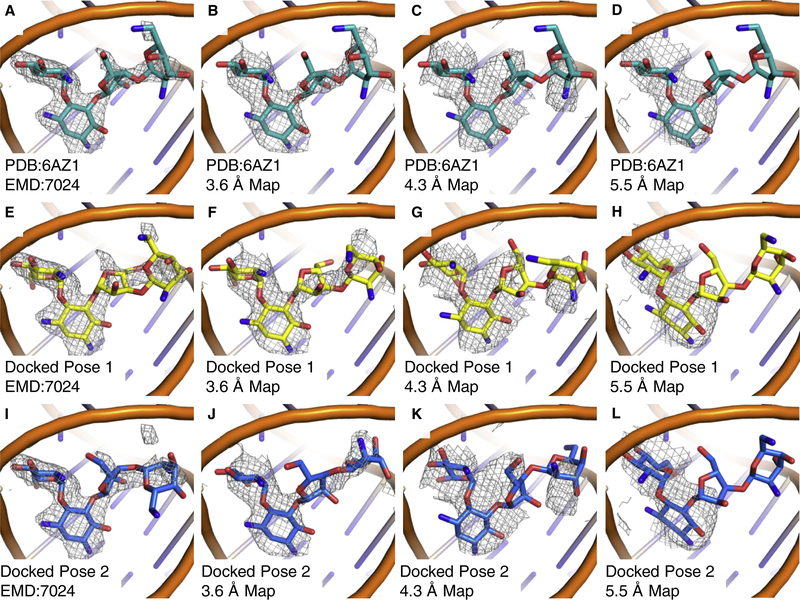
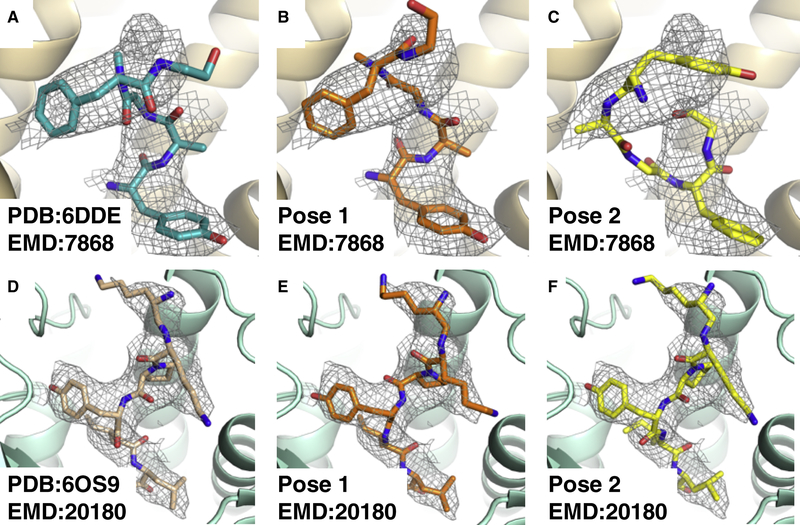
Producing an accurate atomic model of biomolecule-ligand interactions from maps generated by cryoelectron microscopy (cryo-EM) often presents challenges inherent to the methodology and the dynamic nature of ligand binding. Here, we present GemSpot, an automated pipeline of computational chemistry methods that take into account EM map potentials, quantum mechanics energy calculations, and water molecule site prediction to generate candidate poses and provide a measure of the degree of confidence. The pipeline is validated through several published cryo-EM structures of complexes in different resolution ranges and various types of ligands. In all cases, at least one identified pose produced both excellent interactions with the target and agreement with the map. GemSpot will be valuable for the robust identification of ligand poses and drug discovery efforts through cryo-EM.
从冷冻电子显微镜(cryo-EM)生成的图谱中生成生物分子-配体相互作用的精确原子模型通常会带来固有于该方法和配体结合的动态特性的挑战。在这里,我们介绍了 GemSpot,这是一种自动化的计算化学方法管道,它考虑了 EM 图谱势能、量子力学能量计算和水分子位置预测,以生成候选构象并提供置信度的度量。该管道通过几个不同分辨率范围和各种类型配体的复合物的已发表的 cryo-EM 结构进行了验证。在所有情况下,至少有一种鉴定的构象与靶标具有极好的相互作用,并与图谱一致。通过 cryo-EM,GemSpot 将有助于稳健地鉴定配体构象和药物发现工作。