Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, MO 65211, USA.
Biomolecules. 2023 Jan 9;13(1):132. doi: 10.3390/biom13010132.
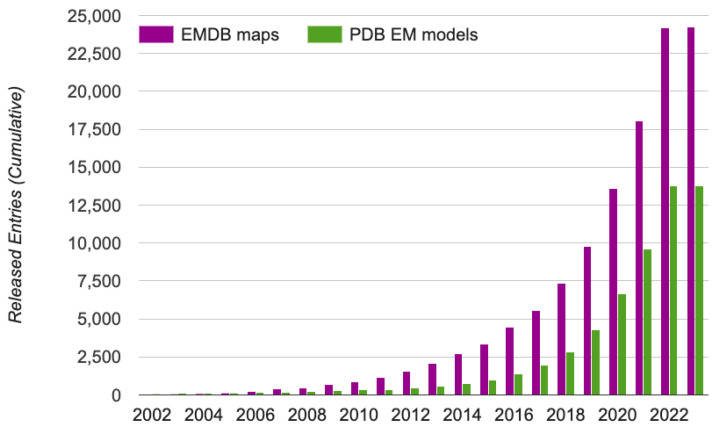
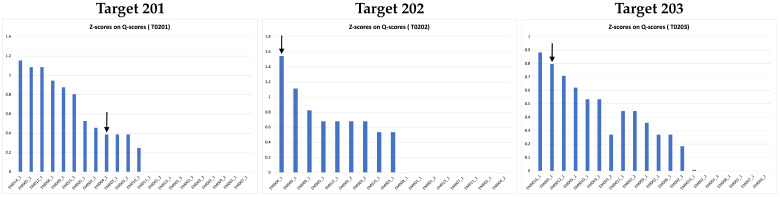
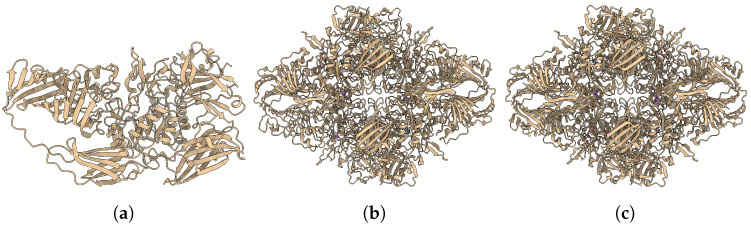
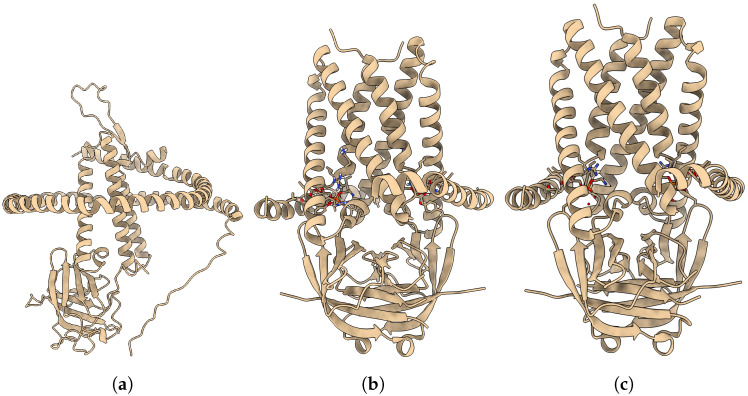
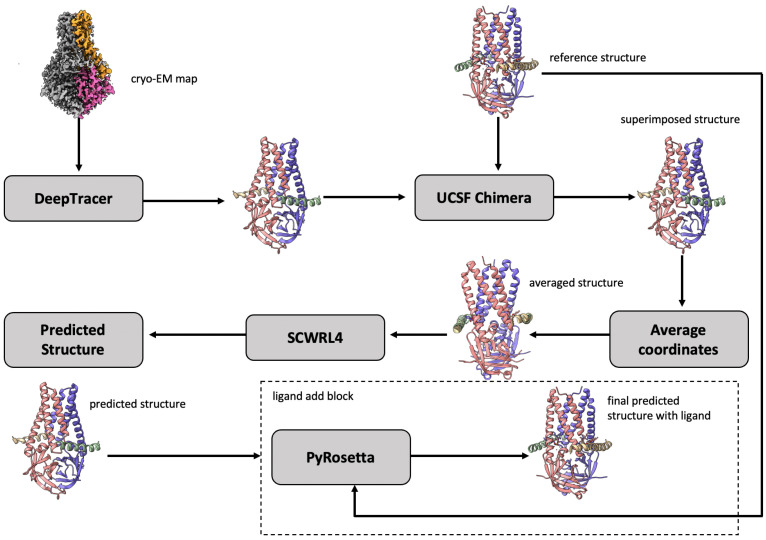
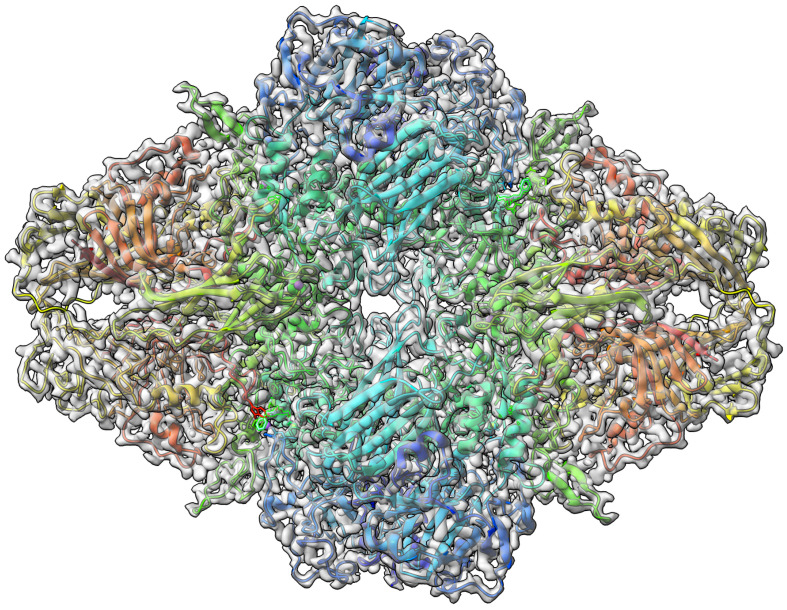
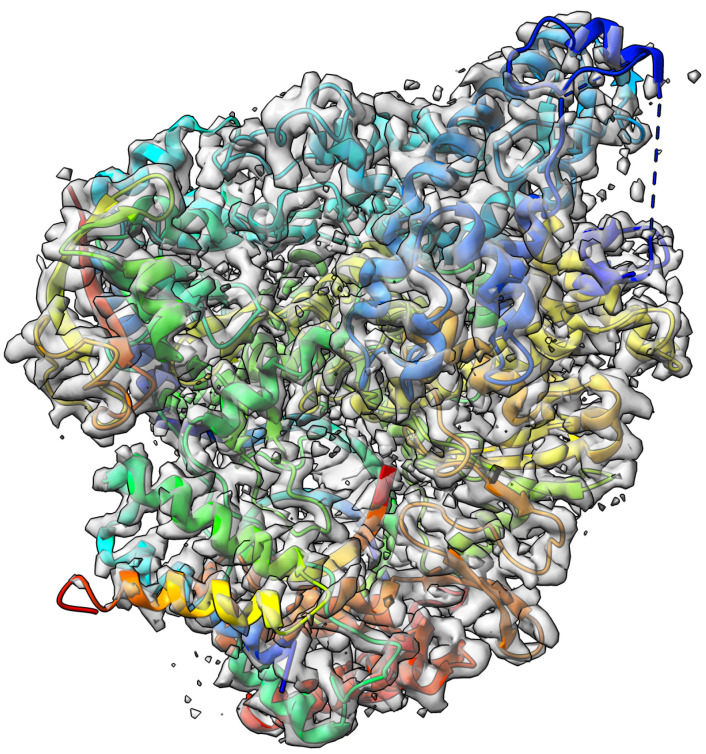
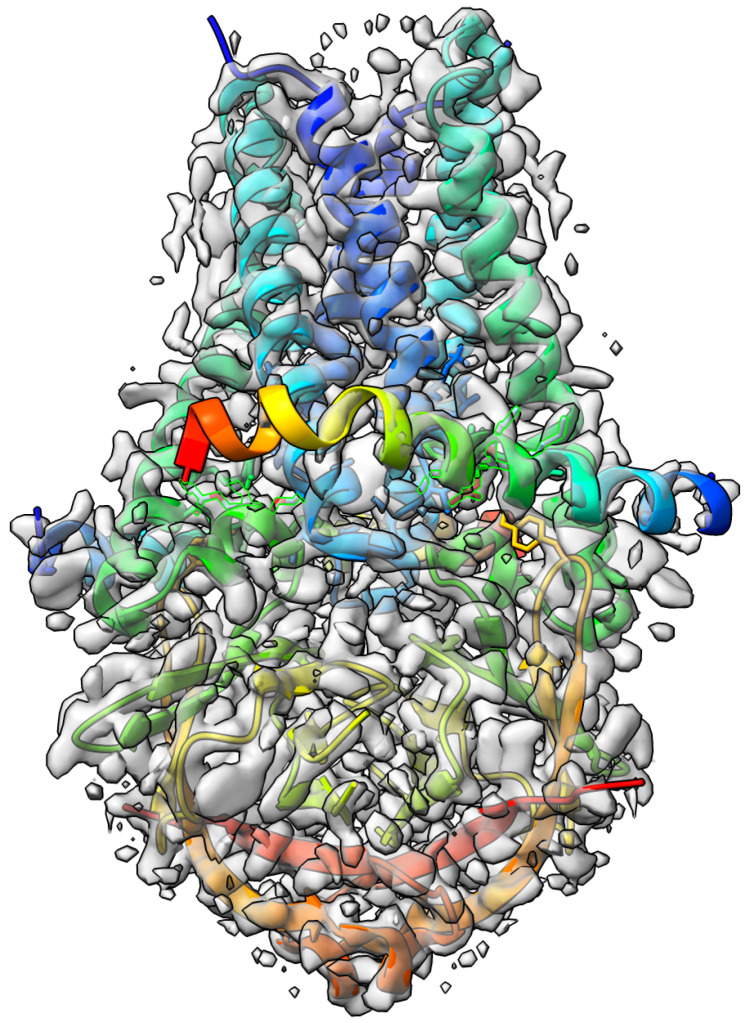
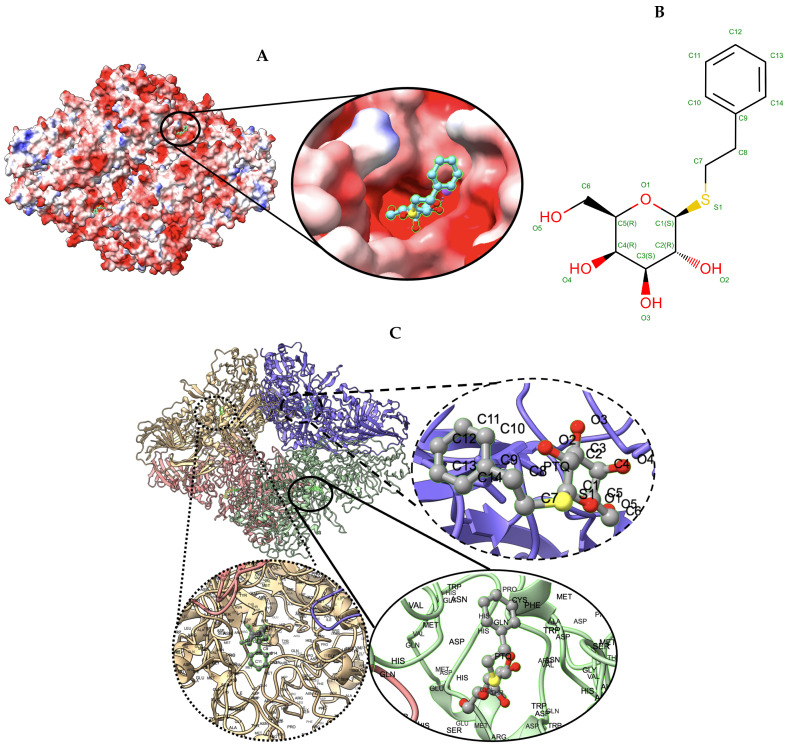
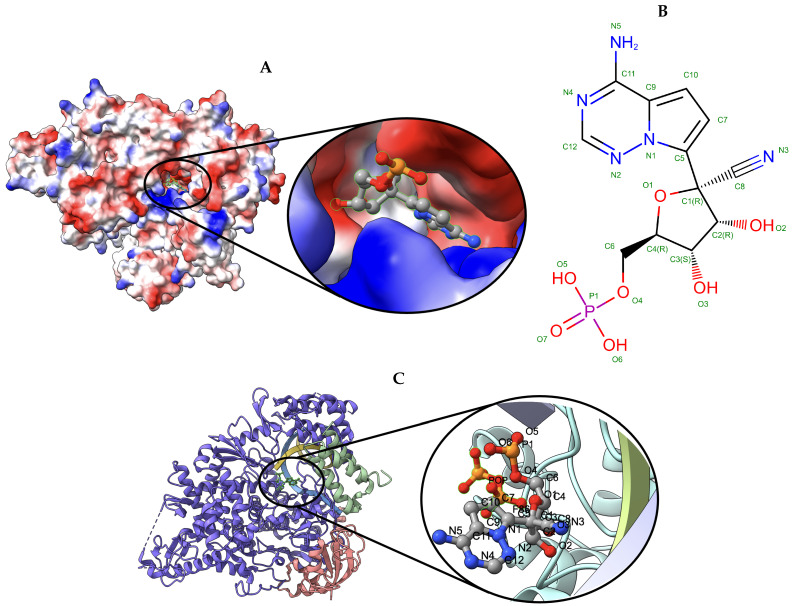
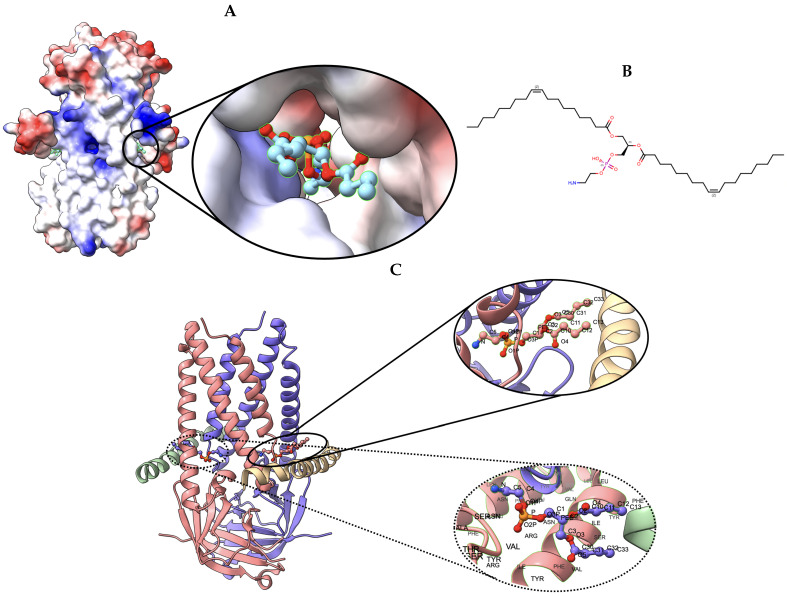
Elucidating protein-ligand interaction is crucial for studying the function of proteins and compounds in an organism and critical for drug discovery and design. The problem of protein-ligand interaction is traditionally tackled by molecular docking and simulation, which is based on physical forces and statistical potentials and cannot effectively leverage cryo-EM data and existing protein structural information in the protein-ligand modeling process. In this work, we developed a deep learning bioinformatics pipeline (DeepProLigand) to predict protein-ligand interactions from cryo-EM density maps of proteins and ligands. DeepProLigand first uses a deep learning method to predict the structure of proteins from cryo-EM maps, which is averaged with a reference (template) structure of the proteins to produce a combined structure to add ligands. The ligands are then identified and added into the structure to generate a protein-ligand complex structure, which is further refined. The method based on the deep learning prediction and template-based modeling was blindly tested in the 2021 EMDataResource Ligand Challenge and was ranked first in fitting ligands to cryo-EM density maps. These results demonstrate that the deep learning bioinformatics approach is a promising direction for modeling protein-ligand interactions on cryo-EM data using prior structural information.
阐明蛋白质-配体相互作用对于研究蛋白质和化合物在生物体中的功能至关重要,对于药物发现和设计也至关重要。传统上,蛋白质-配体相互作用的问题是通过分子对接和模拟来解决的,该方法基于物理力和统计势能,不能有效地利用低温电子显微镜(cryo-EM)数据和蛋白质-配体建模过程中的现有蛋白质结构信息。在这项工作中,我们开发了一个深度学习生物信息学管道(DeepProLigand),用于从蛋白质和配体的 cryo-EM 密度图中预测蛋白质-配体相互作用。DeepProLigand 首先使用深度学习方法从 cryo-EM 图谱中预测蛋白质的结构,然后将其与蛋白质的参考(模板)结构进行平均,生成一个组合结构以添加配体。然后识别配体并将其添加到结构中,生成蛋白质-配体复合物结构,然后对其进行进一步细化。该基于深度学习预测和基于模板建模的方法在 2021 年的 EMDataResource 配体挑战赛中进行了盲测,在拟合配体到 cryo-EM 密度图方面排名第一。这些结果表明,基于深度学习的方法是利用先前的结构信息在 cryo-EM 数据上建模蛋白质-配体相互作用的一个很有前途的方向。