School of Biological Sciences, University of Adelaide, Adelaide, South Australia, Australia.
Department of Biology, Stanford University, Stanford, California, United States of America.
PLoS Genet. 2020 May 29;16(5):e1008827. doi: 10.1371/journal.pgen.1008827. eCollection 2020 May.
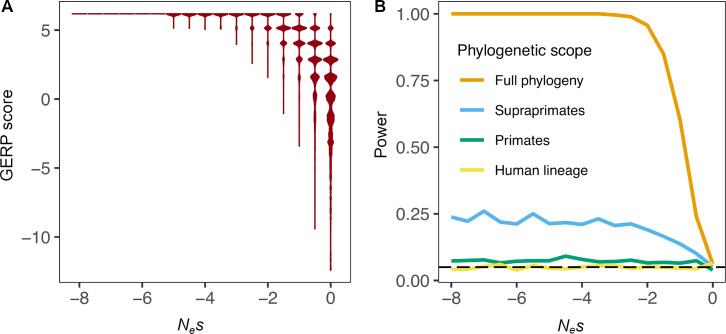
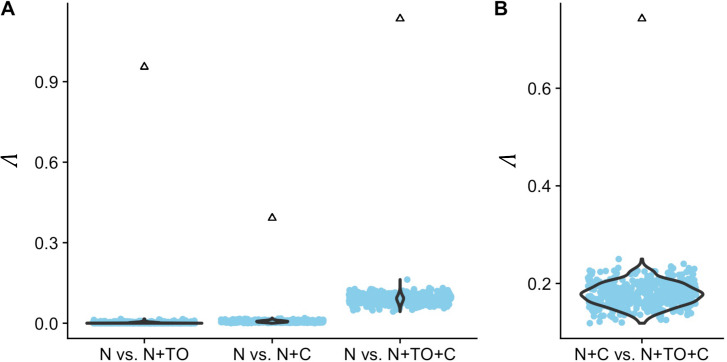
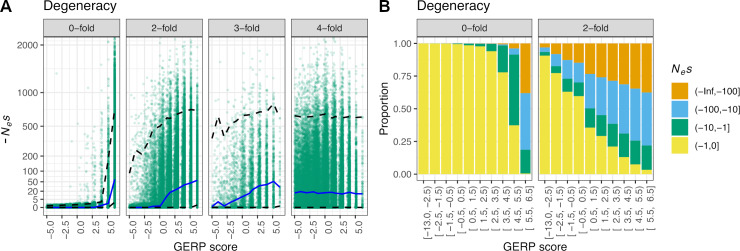
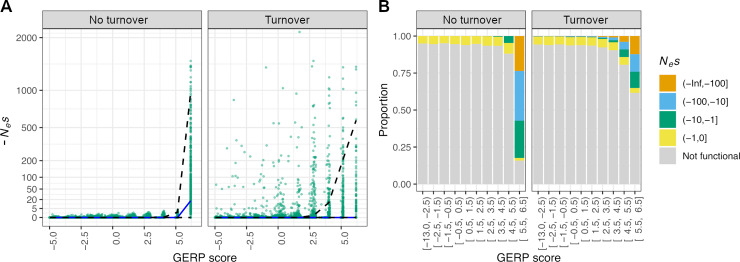
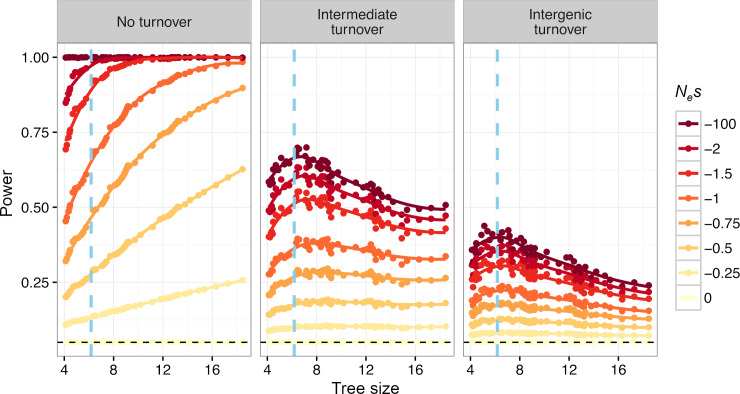
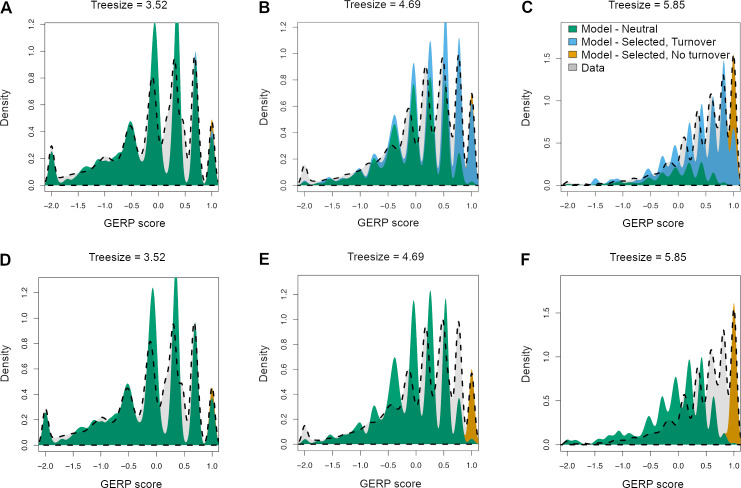
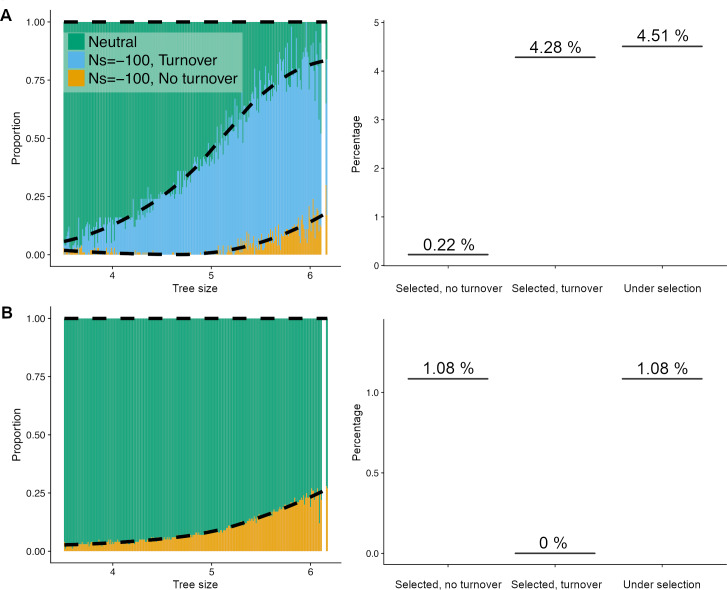
Comparative genomic approaches have been used to identify sites where mutations are under purifying selection and of functional consequence by searching for sequences that are conserved across distantly related species. However, the performance of these approaches has not been rigorously evaluated under population genetic models. Further, short-lived functional elements may not leave a footprint of sequence conservation across many species. We use simulations to study how one measure of conservation, the Genomic Evolutionary Rate Profiling (GERP) score, relates to the strength of selection (Nes). We show that the GERP score is related to the strength of purifying selection. However, changes in selection coefficients or functional elements over time (i.e. functional turnover) can strongly affect the GERP distribution, leading to unexpected relationships between GERP and Nes. Further, we show that for functional elements that have a high turnover rate, adding more species to the analysis does not necessarily increase statistical power. Finally, we use the distribution of GERP scores across the human genome to compare models with and without turnover of sites where mutations are under purifying selection. We show that mutations in 4.51% of the noncoding human genome are under purifying selection and that most of this sequence has likely experienced changes in selection coefficients throughout mammalian evolution. Our work reveals limitations to using comparative genomic approaches to identify deleterious mutations. Commonly used GERP score thresholds miss over half of the noncoding sites in the human genome where mutations are under purifying selection.
比较基因组学方法已被用于通过搜索在远缘物种中保守的序列来识别受到净化选择和功能影响的突变位点。然而,这些方法在群体遗传模型下的性能尚未得到严格评估。此外,短命的功能元件可能不会在许多物种中留下序列保守的痕迹。我们使用模拟来研究一种保守性度量,即基因组进化率分析(GERP)评分,与选择强度(Nes)之间的关系。我们表明,GERP 评分与净化选择的强度有关。然而,选择系数或功能元件随时间的变化(即功能周转率)会强烈影响 GERP 分布,导致 GERP 与 Nes 之间出现意外的关系。此外,我们表明,对于周转率较高的功能元件,增加分析中的物种数量不一定会增加统计能力。最后,我们使用 GERP 评分在人类基因组中的分布来比较具有和不具有净化选择下突变位点周转率的模型。我们表明,人类基因组中非编码区的 4.51%的突变受到净化选择的影响,并且这部分序列很可能在哺乳动物进化过程中经历了选择系数的变化。我们的工作揭示了使用比较基因组学方法识别有害突变的局限性。常用的 GERP 评分阈值错过了人类基因组中受到净化选择的非编码位点的一半以上。