Liang Julia, Pitsillou Eleni, Karagiannis Chris, Darmawan Kevion K, Ng Ken, Hung Andrew, Karagiannis Tom C
Epigenomic Medicine, Department of Diabetes, Central Clinical School, Monash University, Melbourne, VIC 3004, Australia; School of Science, College of Science, Engineering & Health, RMIT University, VIC 3001, Australia.
School of Science, College of Science, Engineering & Health, RMIT University, VIC 3001, Australia; School of Agriculture & Food, Faculty of Veterinary and Agricultural Sciences, University of Melbourne, Parkville, VIC 3052, Australia.
Comput Biol Chem. 2020 May 28;87:107292. doi: 10.1016/j.compbiolchem.2020.107292.
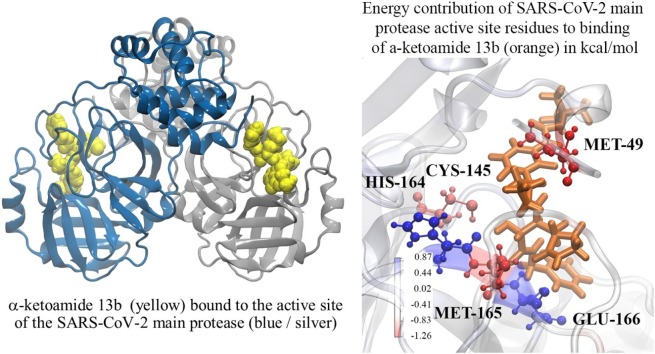
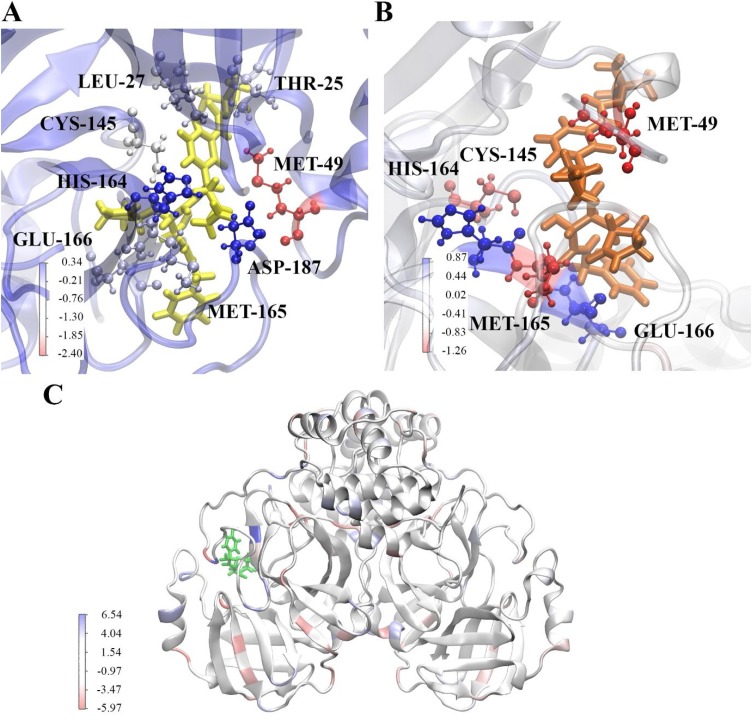
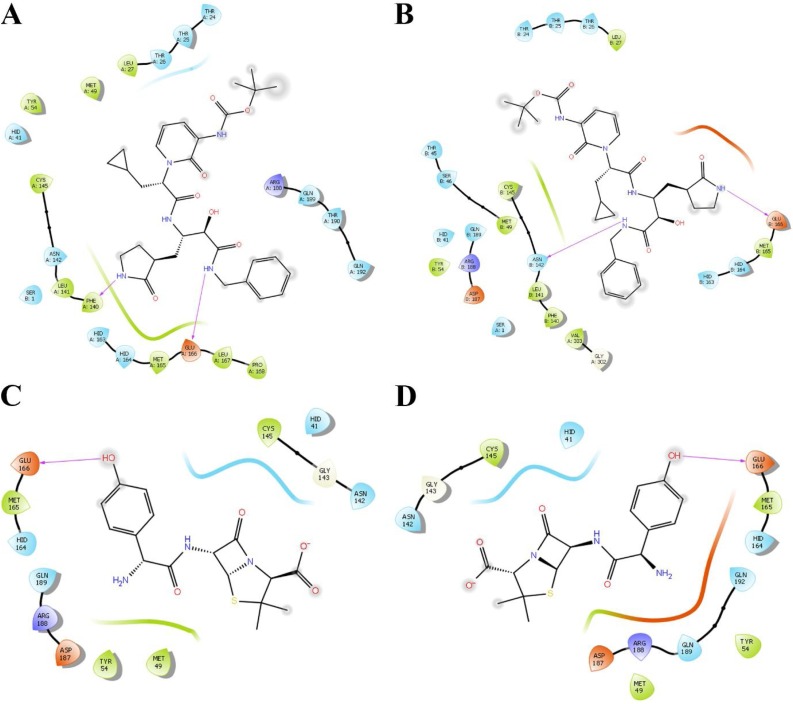
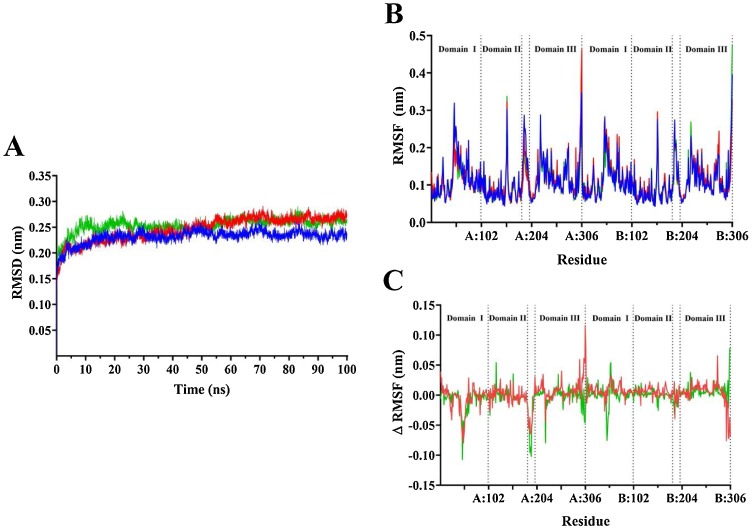
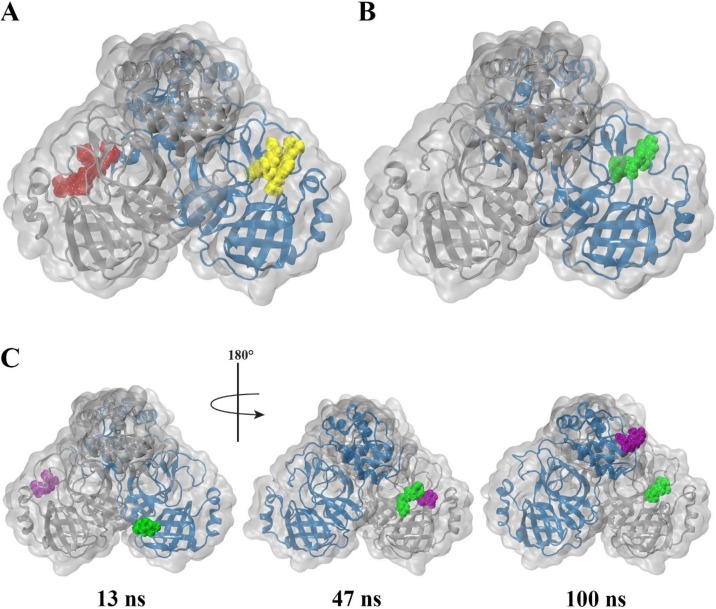
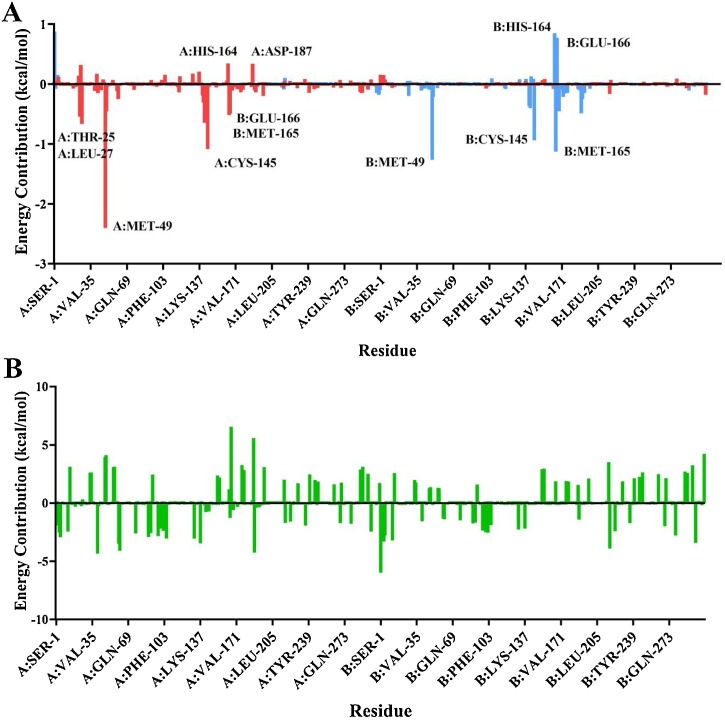
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) causes an illness known as COVID-19, which has been declared a global pandemic with over 2 million confirmed cases and 137,000 deaths in 185 countries and regions at the time of writing (16 April 2020), over a quarter of these cases being in the United States. In the absence of a vaccine, or an approved effective therapeutic, there is an intense interest in repositioning available drugs or designing small molecule antivirals. In this context, in silico modelling has proven to be an invaluable tool. An important target is the SARS-CoV-2 main protease (M), involved in processing translated viral proteins. Peptidomimetic α-ketoamides represent prototypical inhibitors of M. A recent attempt at designing a compound with enhanced pharmacokinetic properties has resulted in the synthesis and evaluation of the α-ketoamide 13b analogue. Here, we performed molecular docking and molecular dynamics simulations to further characterize the interaction of α-ketoamide 13b with the active site of the SARS-CoV-2 M. We included the widely used antibiotic, amoxicillin, for comparison. Our findings indicate that α-ketoamide 13b binds more tightly (predicted GlideScore = -8.7 and -9.2 kcal/mol for protomers A and B, respectively), to the protease active site compared to amoxicillin (-5.0 and -4.8 kcal/mol). Further, molecular dynamics simulations highlight the stability of the interaction of the α-ketoamide 13b ligand with the SARS-CoV-2 M (ΔG = -25.2 and -22.3 kcal/mol for protomers A and B). In contrast, amoxicillin interacts unfavourably with the protease (ΔG = +32.8 kcal/mol for protomer A), with unbinding events observed in several independent simulations. Overall, our findings are consistent with those previously observed, and highlight the need to further explore the α-ketoamides as potential antivirals for this ongoing COVID-19 pandemic.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引发了一种名为COVID-19的疾病,在撰写本文时(2020年4月16日),该疾病已被宣布为全球大流行,在185个国家和地区有超过200万确诊病例和13.7万例死亡,其中超过四分之一的病例在美国。在缺乏疫苗或经批准的有效治疗方法的情况下,人们对重新利用现有药物或设计小分子抗病毒药物有着浓厚兴趣。在这种背景下,计算机模拟已被证明是一种非常有价值的工具。一个重要靶点是SARS-CoV-2主要蛋白酶(M),它参与处理翻译后的病毒蛋白。拟肽α-酮酰胺是M的典型抑制剂。最近一次设计具有增强药代动力学性质化合物的尝试导致了α-酮酰胺13b类似物的合成和评估。在此,我们进行了分子对接和分子动力学模拟,以进一步表征α-酮酰胺13b与SARS-CoV-2 M活性位点的相互作用。我们纳入了广泛使用的抗生素阿莫西林作为对照。我们的研究结果表明,与阿莫西林(分别为-5.0和-4.8 kcal/mol)相比,α-酮酰胺13b与蛋白酶活性位点的结合更紧密(预测的GlideScore值,原聚体A和B分别为-8.7和-9.2 kcal/mol)。此外,分子动力学模拟突出了α-酮酰胺13b配体与SARS-CoV-2 M相互作用的稳定性(原聚体A和B的ΔG分别为-25.2和-22.3 kcal/mol)。相比之下,阿莫西林与蛋白酶的相互作用不利(原聚体A的ΔG为+32.8 kcal/mol),在几个独立模拟中观察到了解离事件。总体而言,我们的研究结果与之前观察到的结果一致,并强调有必要进一步探索α-酮酰胺作为当前COVID-19大流行潜在抗病毒药物的可能性。