National Key Laboratory of Crop Genetic Improvement and Hubei Key Laboratory of Plant Pathology, Huazhong Agricultural University, No.1 Shizishan Street, Hongshan District, Wuhan, 430070, China.
Microbiome. 2020 Jun 3;8(1):80. doi: 10.1186/s40168-020-00859-0.
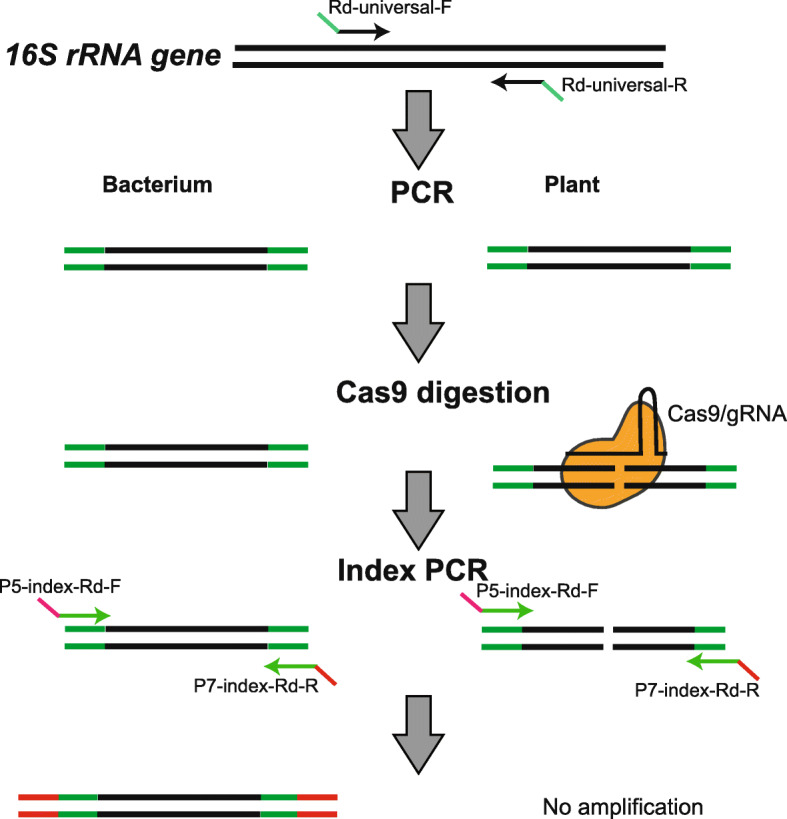
High-throughput sequencing of bacterial 16S rRNA gene (16S-seq) is a useful and common method for studying bacterial community structures. However, contamination of the 16S rRNA genes from the mitochondrion and plastid hinders the sensitive bacterial 16S-seq in plant microbiota profiling, especially for some plant species such as rice. To date, efficiently mitigating such host contamination without a bias is challenging in 16S rRNA gene-based amplicon sequencing.
We developed Cas-16S-seq method to reduce abundant host contamination for plant microbiota profiling. This method utilizes the Cas9 nuclease and specific guide RNA (gRNA) to cut 16S rRNA targets during library construction, thereby removing host contamination in 16S-seq. We used rice as an example to validate the feasibility and effectiveness of Cas-16S-seq. We established a bioinformatics pipeline to design gRNAs that specifically target rice 16S rRNA genes without bacterial 16S rRNA off-targets. We compared the effectiveness of Cas-16S-seq with that of the commonly used 16S-seq method for artificially mixed 16S rRNA gene communities, paddy soil, rice root, and phyllosphere samples. The results showed that Cas-16S-seq substantially reduces the fraction of rice 16S rRNA gene sequences from 63.2 to 2.9% in root samples and from 99.4 to 11.6% in phyllosphere samples on average. Consequently, Cas-16S-seq detected more bacterial species than the 16S-seq in plant samples. Importantly, when analyzing soil samples, Cas-16S-seq and 16S-seq showed almost identical bacterial communities, suggesting that Cas-16S-seq with host-specific gRNAs that we designed has no off-target in rice microbiota profiling.
Our Cas-16S-seq can efficiently remove abundant host contamination without a bias for 16S rRNA gene-based amplicon sequencing, thereby enabling deeper bacterial community profiling with a low cost and high flexibility. Thus, we anticipate that this method would be a useful tool for plant microbiomics. Video Abstract.
细菌 16S rRNA 基因高通量测序(16S-seq)是研究细菌群落结构的一种有用且常见的方法。然而,线粒体和质体 16S rRNA 基因的污染会阻碍植物微生物组分析中灵敏的细菌 16S-seq,尤其是对于水稻等一些植物物种。迄今为止,在基于 16S rRNA 基因扩增子测序的情况下,有效地减轻这种宿主污染而不产生偏差是具有挑战性的。
我们开发了 Cas-16S-seq 方法来减少植物微生物组分析中的丰富宿主污染。该方法利用 Cas9 核酸酶和特定的向导 RNA(gRNA)在文库构建过程中切割 16S rRNA 靶标,从而去除 16S-seq 中的宿主污染。我们以水稻为例验证了 Cas-16S-seq 的可行性和有效性。我们建立了一个生物信息学管道来设计特异性靶向水稻 16S rRNA 基因而没有细菌 16S rRNA 脱靶的 gRNA。我们比较了 Cas-16S-seq 与常用的 16S-seq 方法在人工混合 16S rRNA 基因群落、稻田土壤、水稻根和叶际样本中的效果。结果表明,Cas-16S-seq 可使根样本中水稻 16S rRNA 基因序列的比例从 63.2%降至 2.9%,叶际样本中从 99.4%降至 11.6%,平均降幅达 70%。因此,Cas-16S-seq 在植物样本中检测到的细菌种类多于 16S-seq。重要的是,在分析土壤样本时,Cas-16S-seq 和 16S-seq 显示出几乎相同的细菌群落,这表明我们设计的基于宿主特异性 gRNA 的 Cas-16S-seq 在水稻微生物组分析中没有脱靶。
我们的 Cas-16S-seq 可以有效地去除基于 16S rRNA 基因扩增子测序的丰富宿主污染而不产生偏差,从而以低成本和高灵活性实现更深入的细菌群落分析。因此,我们预计该方法将成为植物微生物组学的有用工具。