School of Medicine, Xiamen University, Xiamen, China.
Department of Intensive Care Unit, The First Affiliated Hospital of Xiamen University, Xiamen, China.
Front Endocrinol (Lausanne). 2020 May 20;11:272. doi: 10.3389/fendo.2020.00272. eCollection 2020.
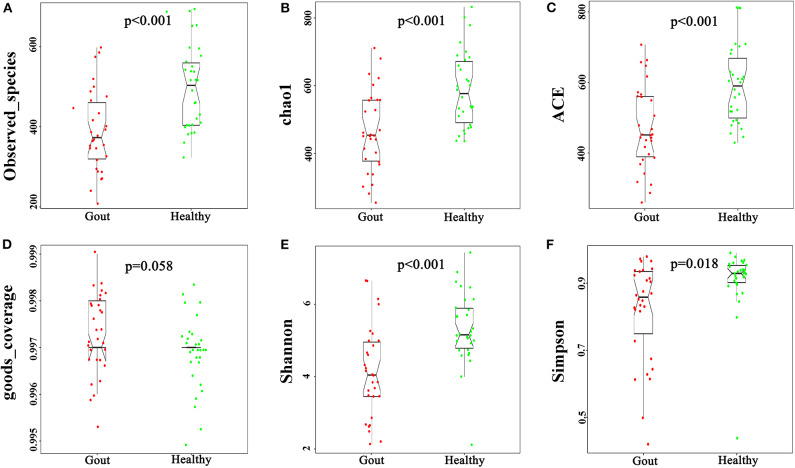
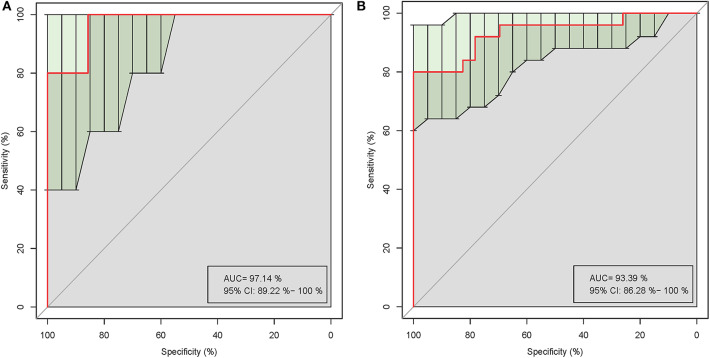
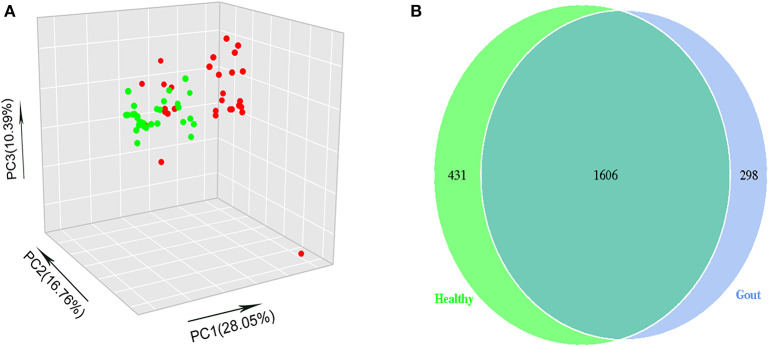
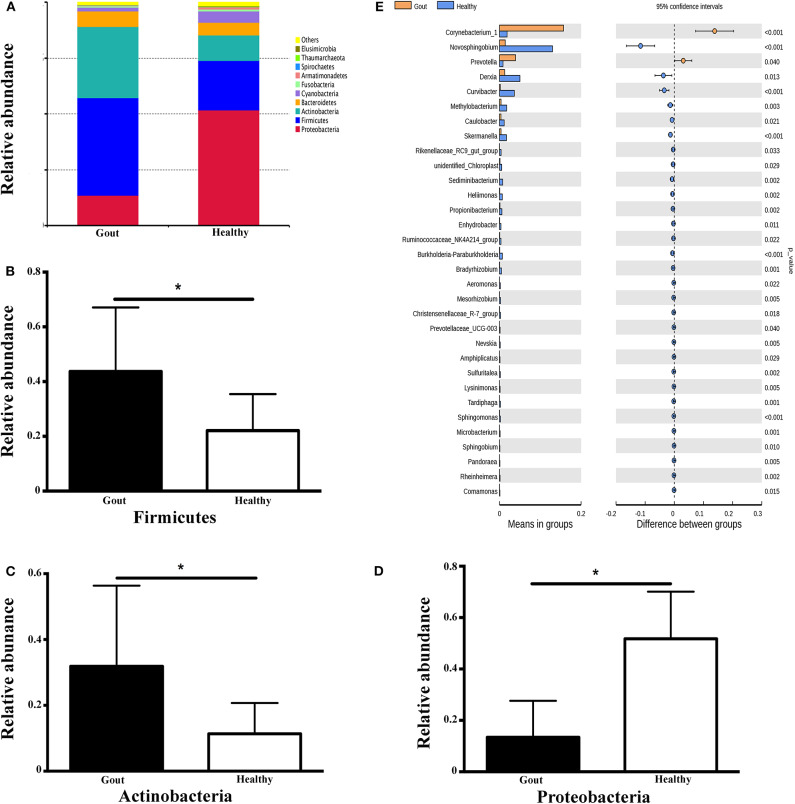
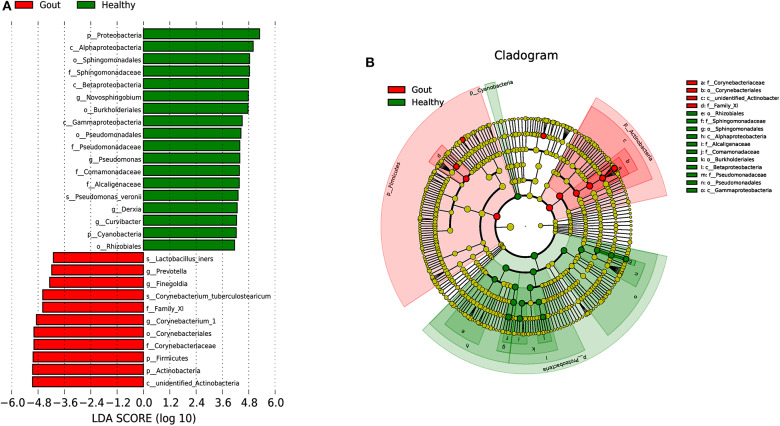
The role of host microbes in the pathogenesis of several diseases has been established, and altered microbiomes have been related to diseases. However, the variability of the urinary microbiome in individuals with gout has not been evaluated to date. Therefore, we conducted the present prospective study to characterize the urinary microbiome and its potential relation to gout. Urine samples from 30 patients with gout and 30 healthy controls were analyzed by Illumina MiSeq sequencing of the 16S rRNA hypervariable regions, and the microbiomes were compared according to alpha-diversity indices, complexity (beta diversity) with principal component analysis, and composition with linear discriminant analysis effect size. The most significantly different taxa at the phylum and genus levels were identified, and their potential as biomarkers for discriminating gout patients was assessed based on receiver operating characteristic (ROC) curve analysis. Compared with the healthy controls, there was a dramatic decrease in microbial richness and diversity in the urine of gout patients. The phylum Firmicutes and its derivatives (Lactobacillus_iners, Family_XI, and Finegoldia), the phylum Actinobacteria and its derivatives (unidentified_Actinobacteria, Corynebacteriales, Corynebacteriale, Corynebacterium_1, and Corynebacterium_tuberculostearicum), and the genera and Corynebacterium_1 were significantly enriched in the urine of gout patients. ROC analysis indicated that the top five altered microbial genera could be reliable markers for distinguishing gout patients from healthy individuals. These findings demonstrate that there are specific alterations in the microbial diversity of gout patients. Thus, further studies on the causal relationship between gout and the urinary microbiome will offer new prospects for diagnosing, preventing, and treating gout.
宿主微生物在几种疾病的发病机制中的作用已经确立,并且微生物组的改变与疾病有关。然而,迄今为止,尚未评估痛风患者尿液微生物组的个体变异性。因此,我们进行了本前瞻性研究,以描述尿液微生物组及其与痛风的潜在关系。通过 Illumina MiSeq 对 16S rRNA 高变区进行测序,分析了 30 例痛风患者和 30 例健康对照者的尿液样本,并根据 alpha 多样性指数、主成分分析的复杂性(beta 多样性)和线性判别分析效应大小比较了微生物组。鉴定了在门和属水平上差异最显著的分类群,并基于接收者操作特征(ROC)曲线分析评估了它们作为区分痛风患者的生物标志物的潜力。与健康对照组相比,痛风患者尿液中的微生物丰富度和多样性显著降低。厚壁菌门及其衍生物(乳杆菌属、未鉴定的厚壁菌门、梭菌科和梭菌目)、放线菌门及其衍生物(未鉴定的放线菌门、棒状杆菌科、棒状杆菌目、棒状杆菌属 1 和结核棒状杆菌)和属和棒状杆菌属 1 在痛风患者的尿液中明显富集。ROC 分析表明,前五个改变的微生物属可以作为区分痛风患者和健康个体的可靠标志物。这些发现表明,痛风患者的微生物多样性存在特定的改变。因此,进一步研究痛风与尿液微生物组之间的因果关系将为痛风的诊断、预防和治疗提供新的前景。