Liu Lian, Lei Xiujuan, Fang Zengqiang, Tang Yujiao, Meng Jia, Wei Zhen
School of Computer Sciences, Shannxi Normal University, Xi'an, China.
Department of Biological Sciences, Xi'an Jiaotong-Liverpool University, Suzhou, China.
Front Genet. 2020 Jun 9;11:545. doi: 10.3389/fgene.2020.00545. eCollection 2020.
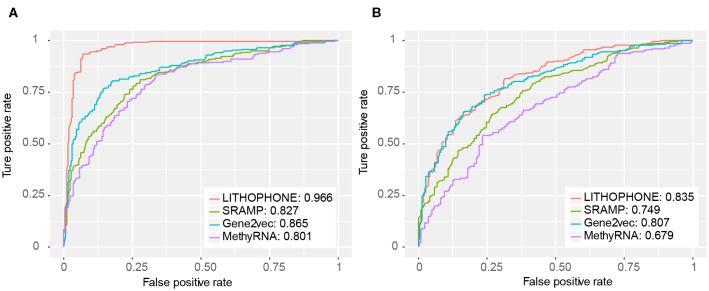
-methyladenosine (mA) is one of the most widely studied epigenetic modifications, which plays an important role in many biological processes, such as splicing, RNA localization, and degradation. Studies have shown that mA on lncRNA has important functions, including regulating the expression and functions of lncRNA, regulating the synthesis of pre-mRNA, promoting the proliferation of cancer cells, and affecting cell differentiation and many others. Although a number of methods have been proposed to predict mA RNA methylation sites, most of these methods aimed at general mA sites prediction without noticing the uniqueness of the lncRNA methylation prediction problem. Since many lncRNAs do not have a polyA tail and cannot be captured in the polyA selection step of the most widely adopted RNA-seq library preparation protocol, lncRNA methylation sites cannot be effectively captured and are thus likely to be significantly underrepresented in existing experimental data affecting the accuracy of existing predictors. In this paper, we propose a new computational framework, , which stands for ong noncodng RNA meylatin sites rediction from sequence caracteristics and genmic iformation with an nsemble predictor. We show that the methylation sites of lncRNA and mRNA have different patterns exhibited in the extracted features and should be differently handled when making predictions. Due to the used experiment protocols, the number of known lncRNA mA sites is limited, and insufficient to train a reliable predictor; thus, the performance can be improved by combining both lncRNA and mRNA data using an ensemble predictor. We show that the newly developed LITHOPHONE approach achieved a reasonably good performance when tested on independent datasets (AUC: 0.966 and 0.835 under full transcript and mature mRNA modes, respectively), marking a substantial improvement compared with existing methods. Additionally, LITHOPHONE was applied to scan the entire human lncRNAome for all possible lncRNA mA sites, and the results are freely accessible at: http://180.208.58.19/lith/.
N6-甲基腺苷(mA)是研究最为广泛的表观遗传修饰之一,它在许多生物学过程中发挥着重要作用,如剪接、RNA定位和降解。研究表明,长链非编码RNA(lncRNA)上的mA具有重要功能,包括调节lncRNA的表达和功能、调控前体mRNA的合成、促进癌细胞增殖以及影响细胞分化等诸多方面。尽管已经提出了许多方法来预测mA RNA甲基化位点,但这些方法大多旨在预测一般的mA位点,而没有注意到lncRNA甲基化预测问题的独特性。由于许多lncRNA没有polyA尾,无法在最广泛采用的RNA测序文库制备方案的polyA选择步骤中被捕获,lncRNA甲基化位点无法被有效捕获,因此在现有实验数据中可能显著代表性不足,影响了现有预测器的准确性。在本文中,我们提出了一种新的计算框架LITHOPHONE,它代表基于序列特征和基因组信息并结合集成预测器的长链非编码RNA甲基化位点预测。我们表明,lncRNA和mRNA的甲基化位点在提取的特征中表现出不同的模式,在进行预测时应区别对待。由于所使用的实验方案,已知的lncRNA mA位点数量有限,不足以训练出一个可靠的预测器;因此,通过使用集成预测器结合lncRNA和mRNA数据可以提高性能。我们表明,新开发的LITHOPHONE方法在独立数据集上测试时取得了相当不错的性能(在完整转录本和成熟mRNA模式下,AUC分别为0.966和0.835),与现有方法相比有了显著改进。此外,LITHOPHONE被应用于扫描整个人类lncRNA组以寻找所有可能的lncRNA mA位点,结果可在以下网址免费获取:http://180.208.58.19/lith/ 。