Department of Pharmaceutical Chemistry, University of Vienna, Althanstraße 14, A-1090, Vienna, Austria.
Mol Inform. 2020 Oct;39(10):e2000090. doi: 10.1002/minf.202000090. Epub 2020 Jul 28.
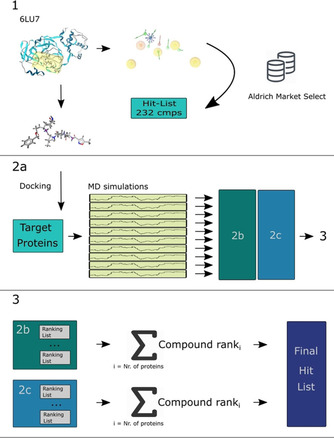
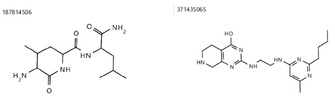
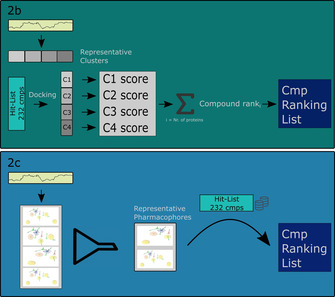
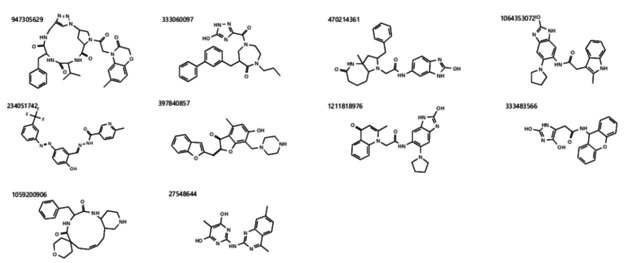
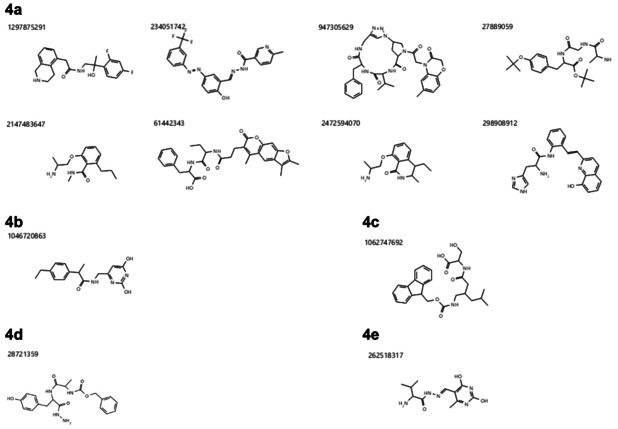
The current pandemic threat of COVID-19, caused by the novel coronavirus SARS-CoV-2, not only gives rise to a high number of deaths around the world but also has immense consequences for the worldwide health systems and global economy. Given the fact that this pandemic is still ongoing and there are currently no drugs or vaccines against this novel coronavirus available, this in silico study was conducted to identify a potential novel SARS-CoV-2-inhibitor. Two different approaches were pursued: 1) The Docking Consensus Approach (DCA) is a novel approach, which combines molecular dynamics simulations with molecular docking. 2) The Common Hits Approach (CHA) in contrast focuses on the combination of the feature information of pharmacophore modeling and the flexibility of molecular dynamics simulations. The application of both methods resulted in the identification of 10 compounds with high coronavirus inhibition potential.
当前由新型冠状病毒 SARS-CoV-2 引起的 COVID-19 大流行不仅在全球范围内导致了大量死亡,而且对全球卫生系统和全球经济也产生了巨大影响。鉴于这种大流行仍在继续,目前尚无针对这种新型冠状病毒的药物或疫苗,因此进行了这项计算机模拟研究以鉴定一种潜在的新型 SARS-CoV-2 抑制剂。采用了两种不同的方法:1)对接共识方法(DCA)是一种新方法,它将分子动力学模拟与分子对接相结合。2)相反,常见命中方法(CHA)侧重于组合药效团建模的特征信息和分子动力学模拟的灵活性。这两种方法的应用都确定了具有高冠状病毒抑制潜力的 10 种化合物。