Department of Public Health Sciences, Penn State College of Medicine, Hershey, PA 17033, USA.
Int J Mol Sci. 2020 Aug 12;21(16):5797. doi: 10.3390/ijms21165797.
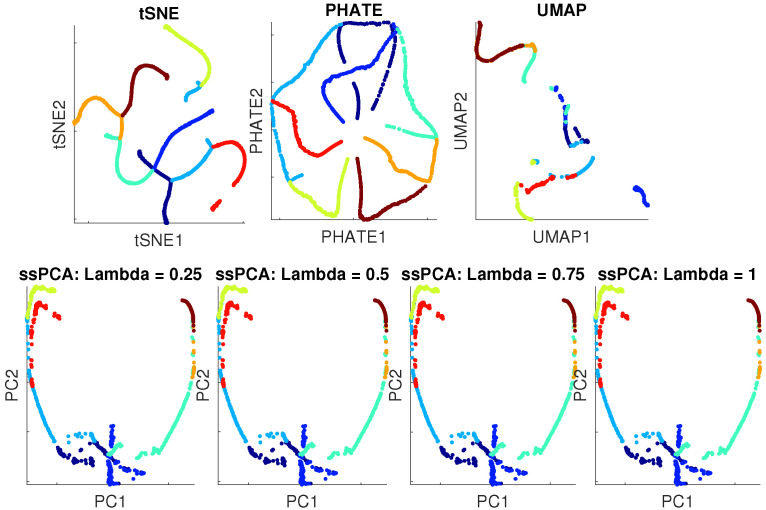
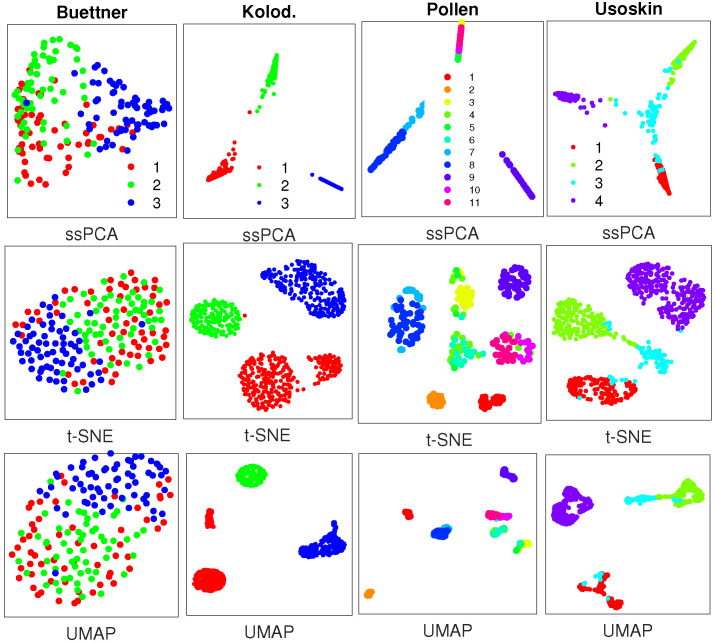
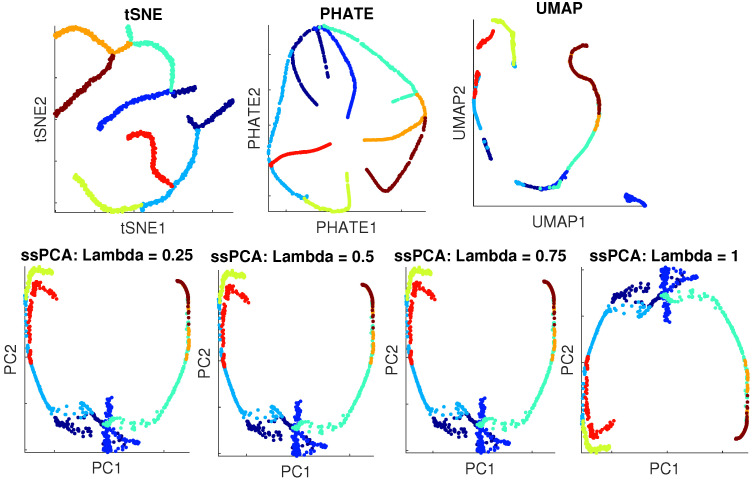
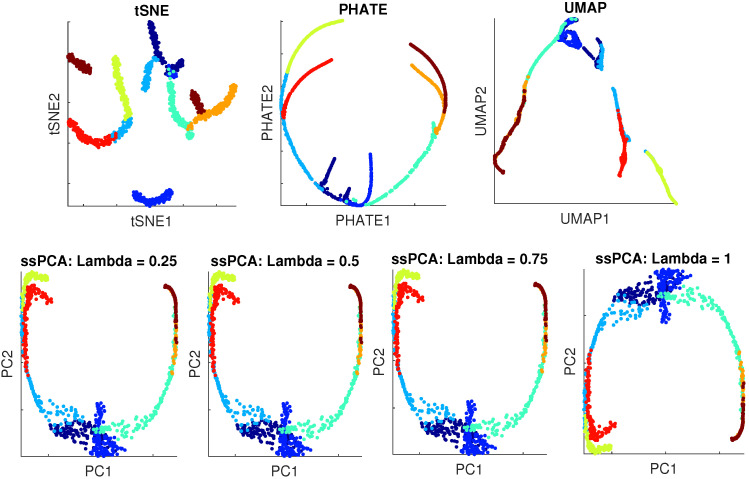
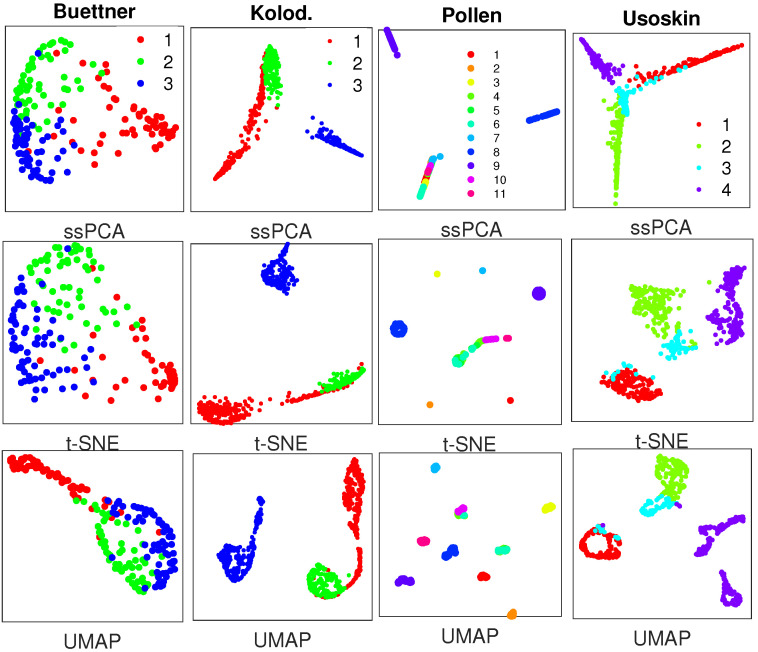
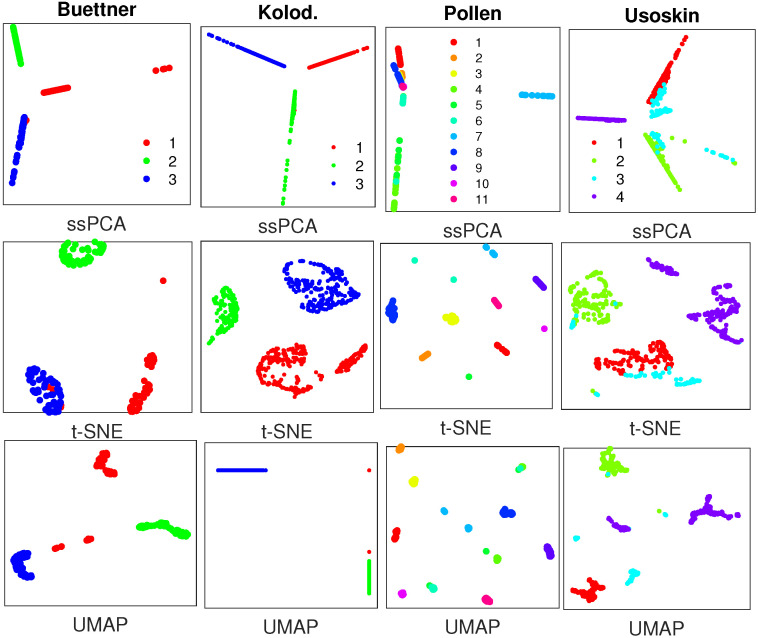
Single-cell RNA-seq (scRNA-seq) is a powerful tool for analyzing heterogeneous and functionally diverse cell population. Visualizing scRNA-seq data can help us effectively extract meaningful biological information and identify novel cell subtypes. Currently, the most popular methods for scRNA-seq visualization are principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE). While PCA is an unsupervised dimension reduction technique, t-SNE incorporates cluster information into pairwise probability, and then maximizes the Kullback-Leibler divergence. Uniform Manifold Approximation and Projection (UMAP) is another recently developed visualization method similar to t-SNE. However, one limitation with UMAP and t-SNE is that they can only capture the local structure of the data, the global structure of the data is not faithfully preserved. In this manuscript, we propose a semisupervised principal component analysis (ssPCA) approach for scRNA-seq visualization. The proposed approach incorporates cluster-labels into dimension reduction and discovers principal components that maximize both data variance and cluster dependence. ssPCA must have cluster-labels as its input. Therefore, it is most useful for visualizing clusters from a scRNA-seq clustering software. Our experiments with simulation and real scRNA-seq data demonstrate that ssPCA is able to preserve both local and global structures of the data, and uncover the transition and progressions in the data, if they exist. In addition, ssPCA is convex and has a global optimal solution. It is also robust and computationally efficient, making it viable for scRNA-seq cluster visualization.
单细胞 RNA 测序 (scRNA-seq) 是分析异质和功能多样化细胞群体的强大工具。可视化 scRNA-seq 数据可以帮助我们有效地提取有意义的生物学信息并识别新的细胞亚型。目前,scRNA-seq 可视化最流行的方法是主成分分析 (PCA) 和 t 分布随机邻域嵌入 (t-SNE)。虽然 PCA 是一种无监督降维技术,但 t-SNE 将聚类信息纳入成对概率中,然后最大化 Kullback-Leibler 散度。Uniform Manifold Approximation and Projection (UMAP) 是另一种最近开发的类似于 t-SNE 的可视化方法。然而,UMAP 和 t-SNE 的一个局限性是它们只能捕获数据的局部结构,而不能忠实地保留数据的全局结构。在本文中,我们提出了一种用于 scRNA-seq 可视化的半监督主成分分析 (ssPCA) 方法。该方法将聚类标签纳入降维过程中,并发现最大化数据方差和聚类依赖性的主成分。ssPCA 必须以聚类标签作为输入。因此,它最适合用于可视化 scRNA-seq 聚类软件中的聚类。我们使用模拟和真实 scRNA-seq 数据进行的实验表明,ssPCA 能够保留数据的局部和全局结构,如果存在的话,还能够揭示数据的转变和进展。此外,ssPCA 是凸的并且具有全局最优解。它还具有鲁棒性和计算效率,使其适用于 scRNA-seq 聚类可视化。