Department of Oncology, The First Affiliated Hospital of Zhengzhou University, No. 1 Eastern Jianshe Road, Erqi District, Zhengzhou, 450000, Henan, China.
Department of Oncology, The First People's Hospital of Zhengzhou, Zhengzhou, 450004, Henan, China.
BMC Microbiol. 2020 Oct 13;20(1):308. doi: 10.1186/s12866-020-01938-w.
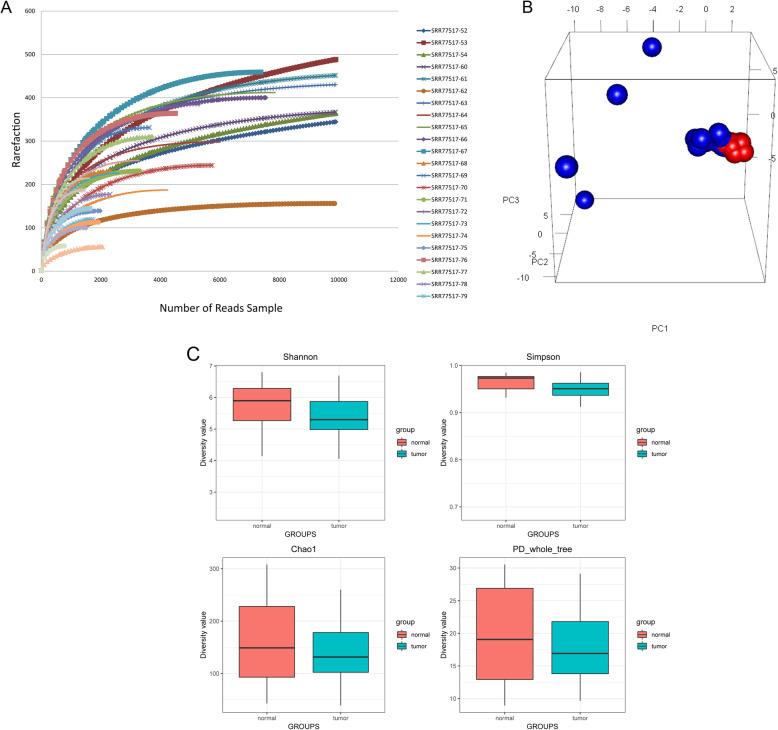
The dysregulation of gut microbiota is pivotal in colorectal carcinogenesis. Meanwhile, altered gut microbiome may affect the development of intestinal diseases through interaction with the host genes. However, the synergy between the altered gut microbiota composition and differential expression of specific genes in colorectal cancer (CRC) remains elusive. Thus, we integrated the data from 16S rRNA gene sequences and RNA sequences to investigate the potential relationship between genes and gut microbes in patients with CRC.
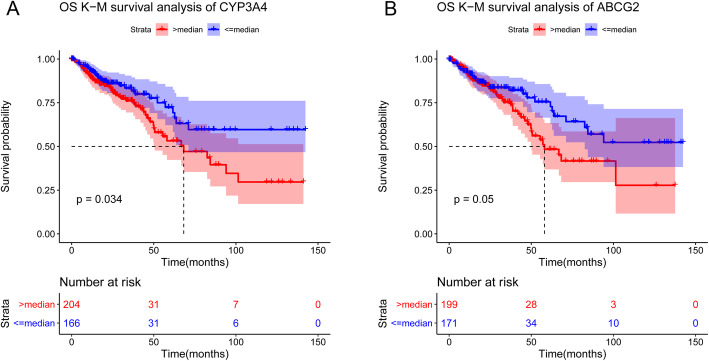
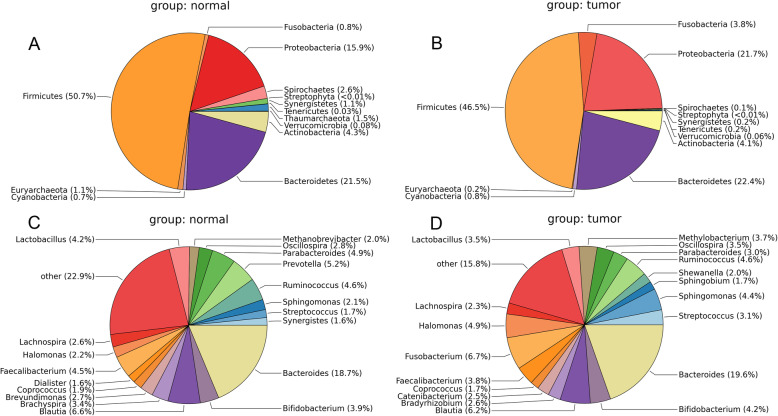
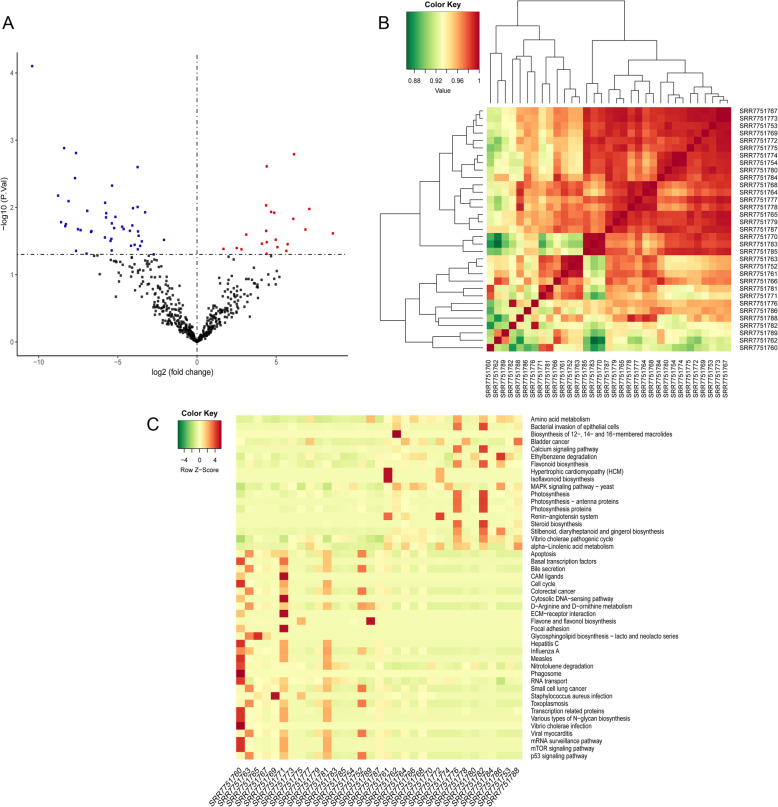
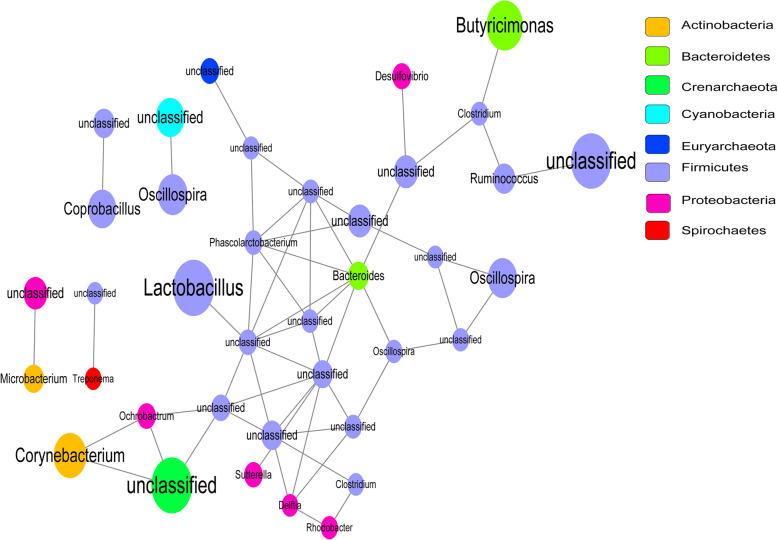
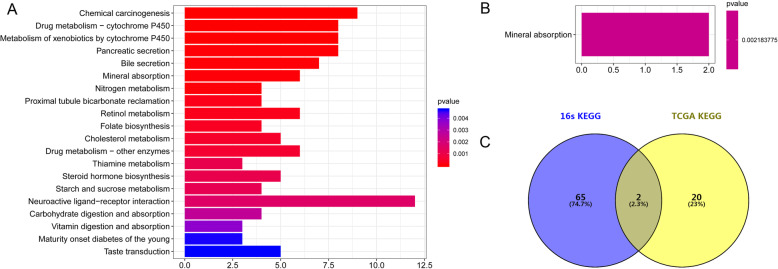
Compared with normal samples, the presence of Proteobacteria and Fusobacteria increased considerably in CRC samples; conversely, the abundance of Firmicutes and Spirochaetes decreased markedly. In particular, the genera Fusobacterium, Catenibacterium, and Shewanella were only detected in tumor samples. Meanwhile, a closely interaction between Butyricimonas and Clostridium was observed in the microbiome network. Furthermore, a total of 246 (differentially expressed genes) DEGs were identified between tumor and normal tissues. Both DEGs and microbiota were involved in bile secretion and steroid hormone biosynthesis pathways. Finally, genes like cytochrome P450 family 3 subfamily A member 4 (CYP3A4) and ATP binding cassette subfamily G member 2 (ABCG2) enriched in these two pathways were connected with the prognosis of CRC, and CRC patients with low expression level of CYP3A4 and ABCG2 had longer survival time.
Identifying the complicated interaction between gut microbiota and the DEGs contributed to further understand the pathogenesis of CRC, and these findings might enable better diagnosis and treatment of CRC patients.
肠道微生物群的失调在结直肠癌的发生中起着关键作用。同时,肠道微生物组的改变可能通过与宿主基因的相互作用影响肠道疾病的发展。然而,改变的肠道微生物群落组成与结直肠癌(CRC)中特定基因的差异表达之间的协同作用仍不清楚。因此,我们整合了 16S rRNA 基因序列和 RNA 序列的数据,以研究 CRC 患者中基因和肠道微生物之间的潜在关系。
与正常样本相比,CRC 样本中变形菌门和梭杆菌门的存在显著增加;相反,厚壁菌门和螺旋体门的丰度明显下降。特别是,梭杆菌属、Catenibacterium 属和希瓦氏菌属仅在肿瘤样本中检测到。同时,在微生物组网络中观察到丁酸单胞菌和梭菌之间的密切相互作用。此外,在肿瘤组织和正常组织之间共鉴定出 246 个(差异表达基因)DEGs。DEGs 和微生物组均参与胆汁分泌和类固醇激素生物合成途径。最后,这些两条途径中富集的细胞色素 P450 家族 3 亚家族 A 成员 4(CYP3A4)和 ATP 结合盒亚家族 G 成员 2(ABCG2)等基因与 CRC 的预后相关,CYP3A4 和 ABCG2 表达水平低的 CRC 患者的生存时间更长。
确定肠道微生物群和差异表达基因之间复杂的相互作用有助于进一步了解 CRC 的发病机制,这些发现可能使 CRC 患者的诊断和治疗得到改善。