Feng Shijing, Liu Zhenshan, Hu Yang, Tian Jieyun, Yang Tuxi, Wei Anzhi
College of Forestry, Northwest A&F University, Yangling, 712100 Shaanxi China.
College of Life Science, Northwest A&F University, Yangling, 712100 Shaanxi China.
Hortic Res. 2020 Oct 1;7:158. doi: 10.1038/s41438-020-00376-z. eCollection 2020.
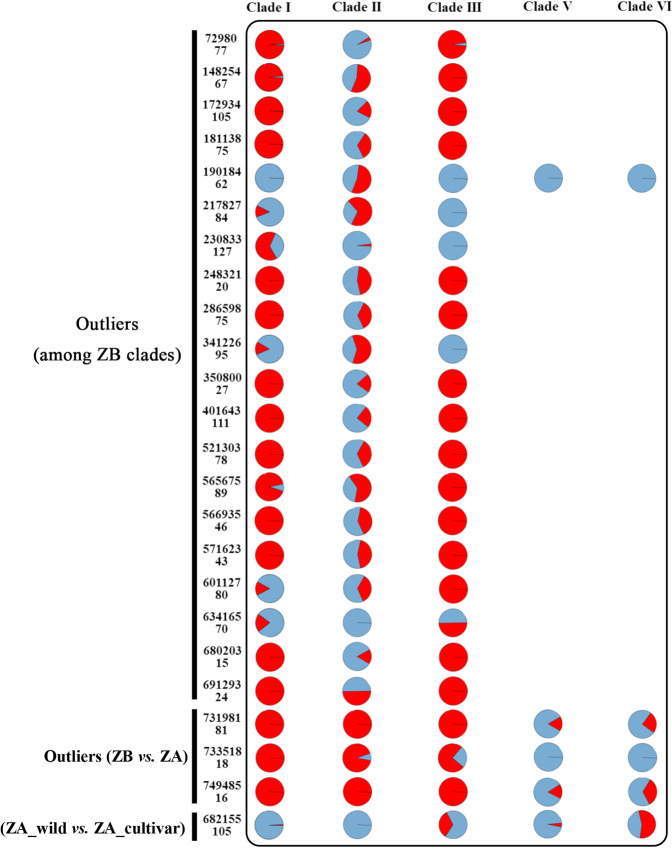
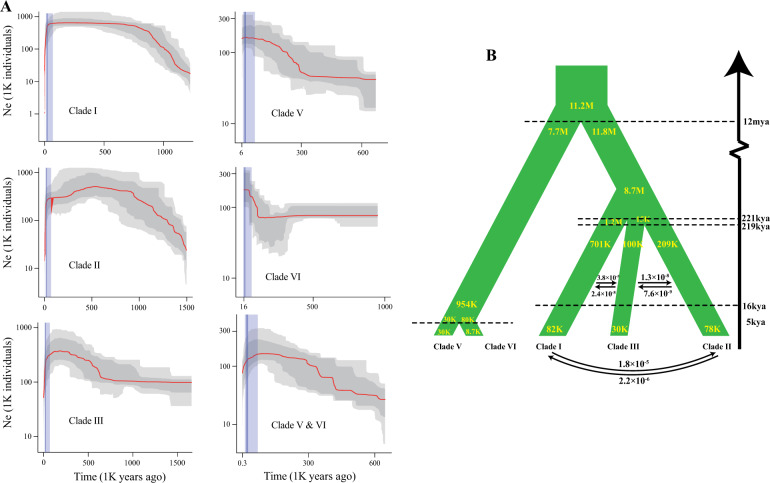
Chinese pepper, mainly including and , is an economically important crop popular in Asian countries due to its unique taste characteristics and potential medical uses. Numerous cultivars of Chinese pepper have been developed in China through long-term domestication. To better understand the population structure, demographic history, and speciation of Chinese pepper, we performed a comprehensive analysis at a genome-wide level by analyzing 38,395 genomic SNPs that were identified in 112 cultivated and wild accessions using a high-throughput genome-wide genotyping-by-sequencing (GBS) approach. Our analysis provides genetic evidence of multiple splitting events occurring between and within species, resulting in at least four clades in and two clades in . Despite no evidence of recent admixture between species, we detected substantial gene flow within species. Estimates of demographic dynamics and species distribution modeling suggest that climatic oscillations during the Pleistocene (including the Penultimate Glaciation and the Last Glacial Maximum) and recent domestication events together shaped the demography and evolution of Chinese pepper. Our analyses also suggest that southeastern Gansu province is the most likely origin of in China. These findings provide comprehensive insights into genetic diversity, population structure, demography, and adaptation in .
花椒,主要包括[具体品种1]和[具体品种2],因其独特的口味特点和潜在的药用价值,是亚洲国家一种具有重要经济价值的作物。在中国,经过长期驯化培育出了众多花椒品种。为了更好地了解花椒的种群结构、种群历史和物种形成,我们采用高通量全基因组简化基因组测序(GBS)方法,对112份栽培和野生种质中鉴定出的38395个基因组单核苷酸多态性(SNP)进行分析,在全基因组水平上进行了全面分析。我们的分析提供了物种间和物种内发生多次分裂事件的遗传证据,导致[品种1]中至少有四个进化枝,[品种2]中有两个进化枝。尽管没有物种间近期杂交的证据,但我们检测到物种内存在大量基因流。种群动态估计和物种分布建模表明,更新世期间的气候振荡(包括倒数第二次冰期和末次盛冰期)以及近期的驯化事件共同塑造了花椒的种群统计学和进化。我们的分析还表明,甘肃省东南部是中国[品种1]最可能的起源地。这些发现为[品种1]的遗传多样性、种群结构、种群统计学和适应性提供了全面的见解。