Xie Haiying, Yang Caiyun, Sun Yamin, Igarashi Yasuo, Jin Tao, Luo Feng
Research Center of Bioenergy and Bioremediation, College of Resources and Environment, Southwest University, Chongqing, China.
PUROTON Gene Medical Institute Co., Ltd., Chongqing, China.
Front Genet. 2020 Sep 8;11:516269. doi: 10.3389/fgene.2020.516269. eCollection 2020.
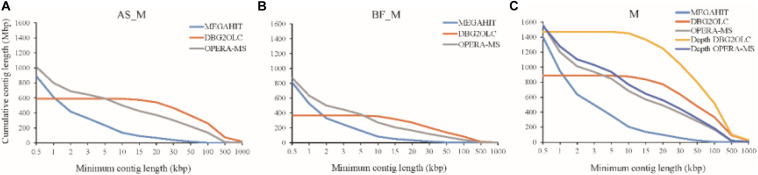
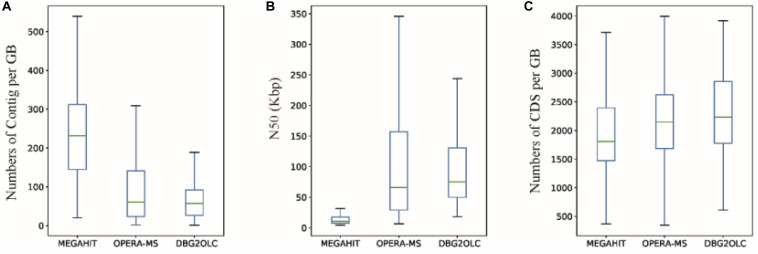
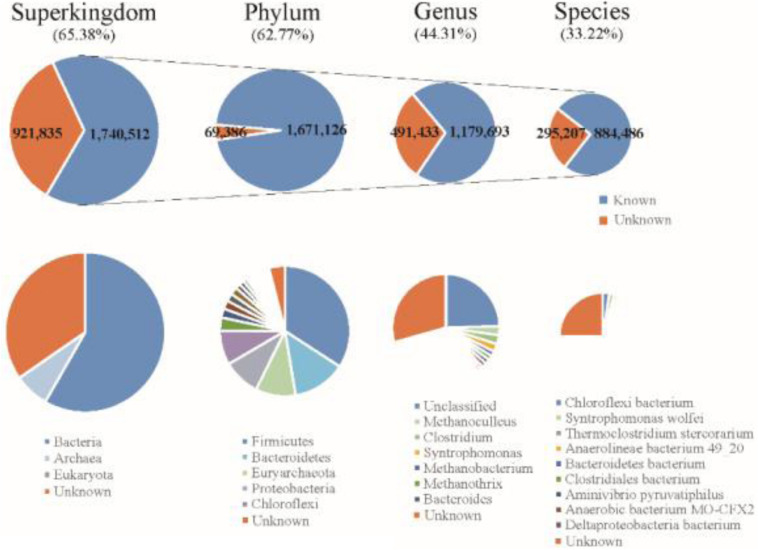
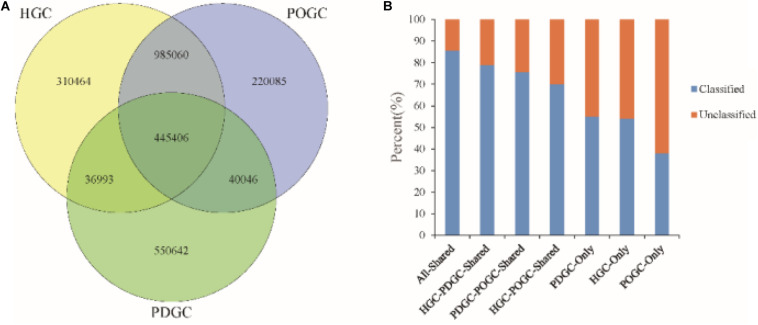
PacBio long reads sequencing presents several potential advantages for DNA assembly, including being able to provide more complete gene profiling of metagenomic samples. However, lower single-pass accuracy can make gene discovery and assembly for low-abundance organisms difficult. To evaluate the application and performance of PacBio long reads and Illumina HiSeq short reads in metagenomic analyses, we directly compared various assemblies involving PacBio and Illumina sequencing reads based on two anaerobic digestion microbiome samples from a biogas fermenter. Using a PacBio platform, 1.58 million long reads (19.6 Gb) were produced with an average length of 7,604 bp. Using an Illumina HiSeq platform, 151.2 million read pairs (45.4 Gb) were produced. Hybrid assemblies using PacBio long reads and HiSeq contigs produced improvements in assembly statistics, including an increase in the average contig length, contig N50 size, and number of large contigs. Interestingly, depth-based hybrid assemblies generated a higher percentage of complete genes (98.86%) compared to those based on HiSeq contigs only (40.29%), because the PacBio reads were long enough to cover many repeating short elements and capture multiple genes in a single read. Additionally, the incorporation of PacBio long reads led to considerable advantages regarding reducing contig numbers and increasing the completeness of the genome reconstruction, which was poorly assembled and binned when using HiSeq data alone. From this comparison of PacBio long reads with Illumina HiSeq short reads related to complex microbiome samples, we conclude that PacBio long reads can produce longer contigs, more complete genes, and better genome binning, thereby offering more information about metagenomic samples.
PacBio长读长测序在DNA组装方面具有几个潜在优势,包括能够提供更完整的宏基因组样本基因谱。然而,较低的单通道准确性会使低丰度生物的基因发现和组装变得困难。为了评估PacBio长读长和Illumina HiSeq短读长在宏基因组分析中的应用和性能,我们基于来自沼气发酵罐的两个厌氧消化微生物组样本,直接比较了涉及PacBio和Illumina测序读长的各种组装。使用PacBio平台,产生了158万个长读长(19.6 Gb),平均长度为7604 bp。使用Illumina HiSeq平台,产生了1.512亿对读段(45.4 Gb)。使用PacBio长读长和HiSeq重叠群进行的混合组装在组装统计方面有改进,包括平均重叠群长度、重叠群N50大小和大重叠群数量的增加。有趣的是,与仅基于HiSeq重叠群的组装(40.29%)相比,基于深度的混合组装产生了更高比例的完整基因(98.86%),因为PacBio读长足够长,可以覆盖许多重复的短元件,并在单个读长中捕获多个基因。此外,纳入PacBio长读长在减少重叠群数量和提高基因组重建完整性方面带来了相当大的优势,仅使用HiSeq数据时,基因组重建组装效果不佳且分类也不好。通过对与复杂微生物组样本相关的PacBio长读长和Illumina HiSeq短读长的比较,我们得出结论,PacBio长读长可以产生更长的重叠群、更完整的基因和更好的基因组分类,从而提供更多关于宏基因组样本的信息。