Department of Mathematical Sciences, University of Copenhagen, Copenhagen, Denmark.
PLoS One. 2020 Oct 29;15(10):e0241405. doi: 10.1371/journal.pone.0241405. eCollection 2020.
The first cases of COVID-19 caused by the SARS-CoV-2 virus were reported in China in December 2019. The disease has since spread globally. Many countries have instated measures to slow the spread of the virus. Information about the spread of the virus in a country can inform the gradual reopening of a country and help to avoid a second wave of infections. Our study focuses on Denmark, which is opening up when this study is performed (end-May 2020) after a lockdown in mid-March.
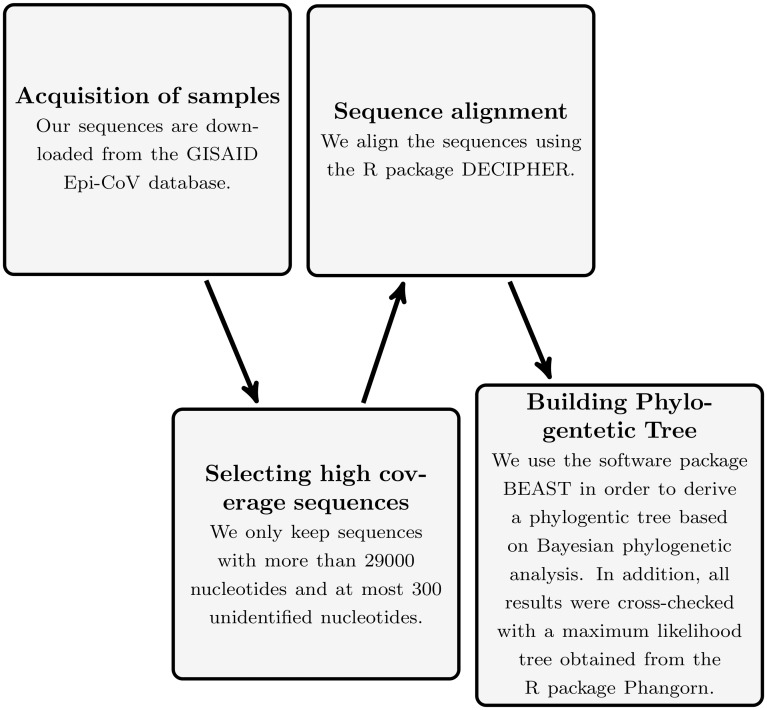
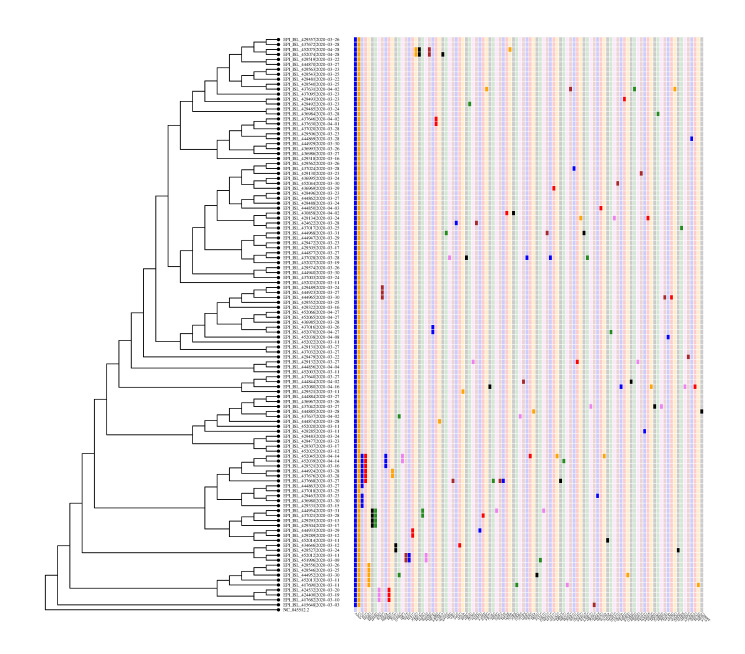
We perform a phylogenetic analysis of 742 publicly available Danish SARS-CoV-2 genome sequences and put them into context using sequences from other countries.
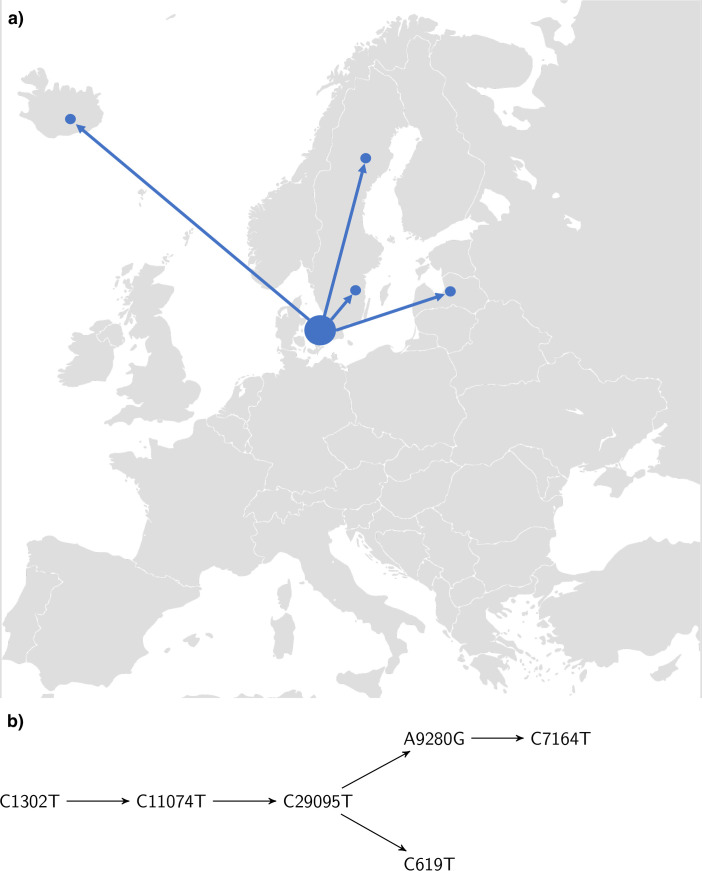
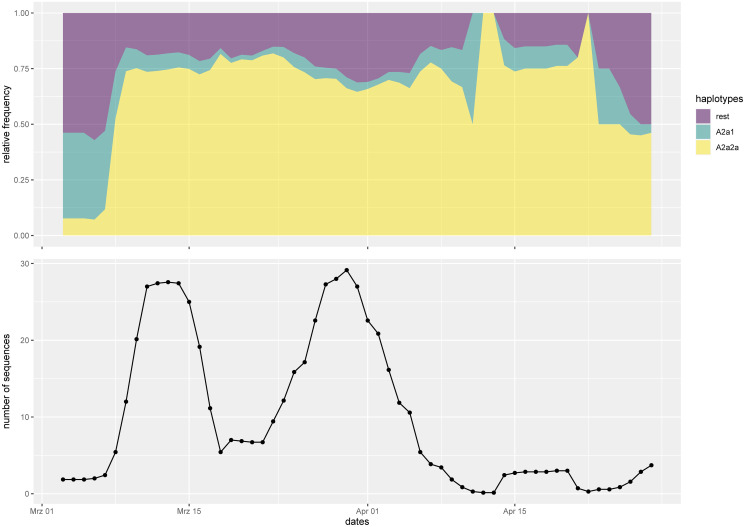
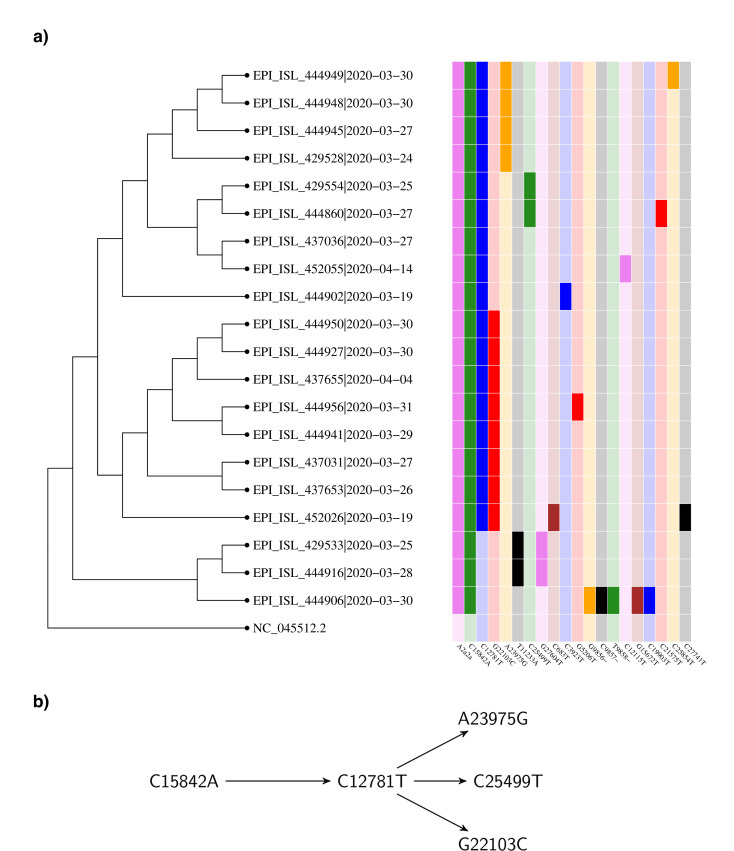
Our findings are consistent with several introductions of the virus to Denmark from independent sources. We identify several chains of mutations that occurred in Denmark. In at least one case we find evidence that the virus spread from Denmark to other countries. A number of the mutations found in Denmark are non-synonymous, and in general there is a considerable variety of strains. The proportions of the most common haplotypes remain stable after lockdown.
Employing phylogenetic methods on Danish genome sequences of SARS-CoV-2, we exemplify how genetic data can be used to trace the introduction of a virus to a country. This provides alternative means for verifying existing assumptions. For example, our analysis supports the hypothesis that the virus was brought to Denmark by skiers returning from Ischgl. On the other hand, we identify transmission routes which suggest that Denmark was part of a network of countries among which the virus was being transmitted. This challenges the common narrative that Denmark only got infected from abroad. Our analysis concerning the ratio of haplotypes does not indicate that the major haplotypes appearing in Denmark have a different degree of virality.
2019 年 12 月,中国首次报告了由严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)引起的 COVID-19 病例。此后,该疾病在全球范围内传播。许多国家都采取措施来减缓病毒的传播。一个国家中病毒传播的信息可以为该国的逐步重新开放提供信息,并有助于避免第二波感染。我们的研究集中在丹麦,该研究在 2020 年 5 月底(进行研究时)进行,此时丹麦在 3 月中旬封锁后正在重新开放。
我们对 742 个公开的丹麦 SARS-CoV-2 基因组序列进行系统发育分析,并使用来自其他国家的序列对其进行分析。
我们的发现与病毒从多个独立来源传入丹麦的情况一致。我们鉴定了在丹麦发生的几个突变链。在至少一个病例中,我们发现了病毒从丹麦传播到其他国家的证据。在丹麦发现的许多突变是非同义突变,并且通常存在相当多的菌株。封锁后,最常见单倍型的比例保持稳定。
我们运用系统发育方法对丹麦 SARS-CoV-2 的基因组序列进行分析,举例说明了如何利用遗传数据来追踪病毒传入一个国家的情况。这为验证现有假设提供了替代方法。例如,我们的分析支持了病毒是由从伊施格尔返回的滑雪者带到丹麦的假设。另一方面,我们发现了传播途径,表明丹麦是病毒在传播的国家网络的一部分。这挑战了丹麦仅从国外感染的常见说法。我们关于单倍型比例的分析表明,出现在丹麦的主要单倍型并没有不同程度的病毒感染力。