Life Sciences Mass Spectrometry, Department of Inorganic and Analytical Chemistry, University of Geneva, 24 Quai Ernest Ansermet, 1211, Geneva 4, Switzerland.
Proteome Informatics Group (PIG), Swiss Institute of Bioinformatics and University of Geneva, 7, route de Drize, 1211, Geneva 4, Switzerland.
Anal Bioanal Chem. 2021 Jan;413(2):503-517. doi: 10.1007/s00216-020-03019-3. Epub 2020 Oct 29.
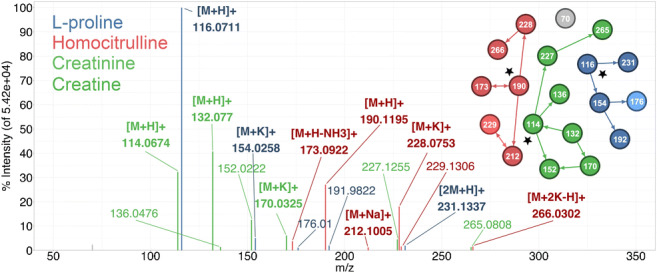
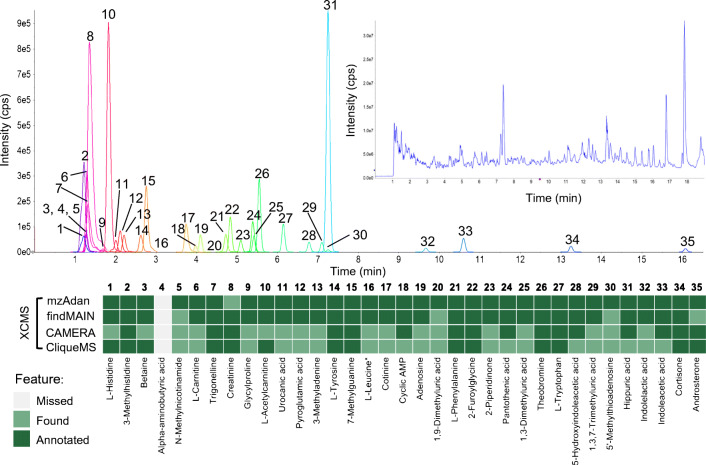
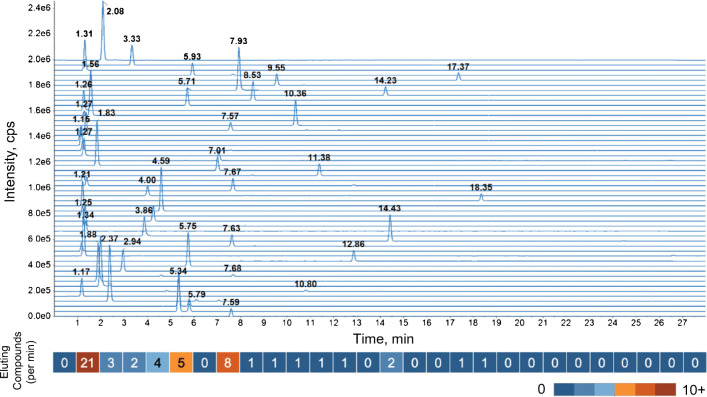
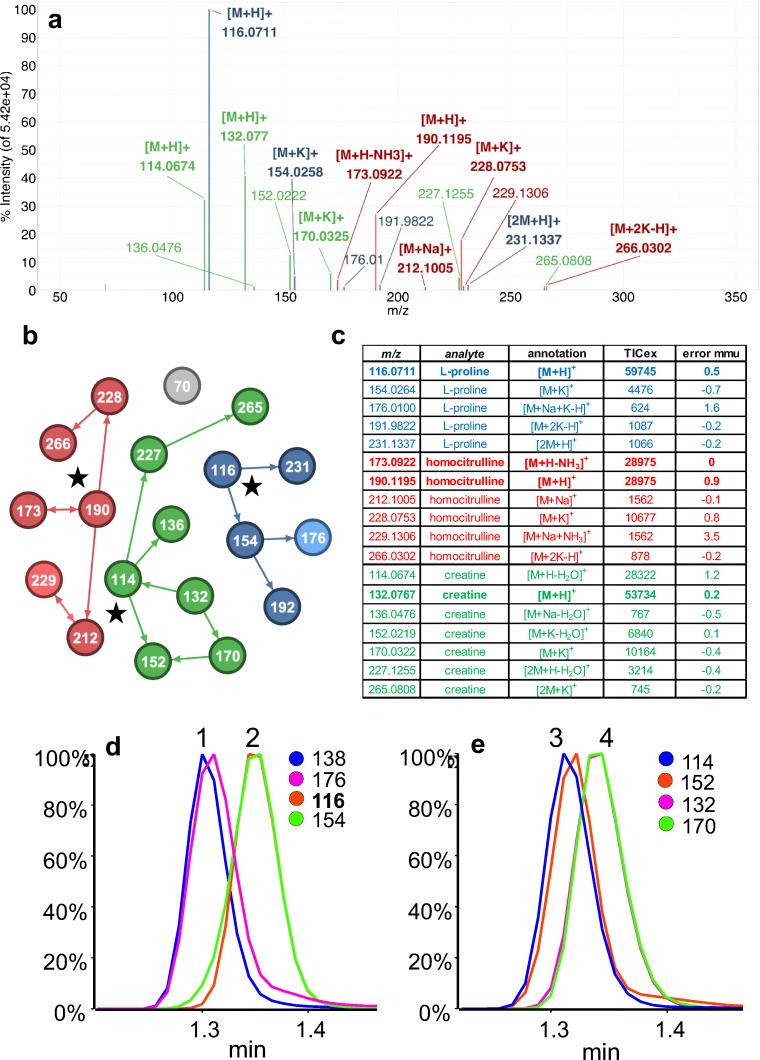
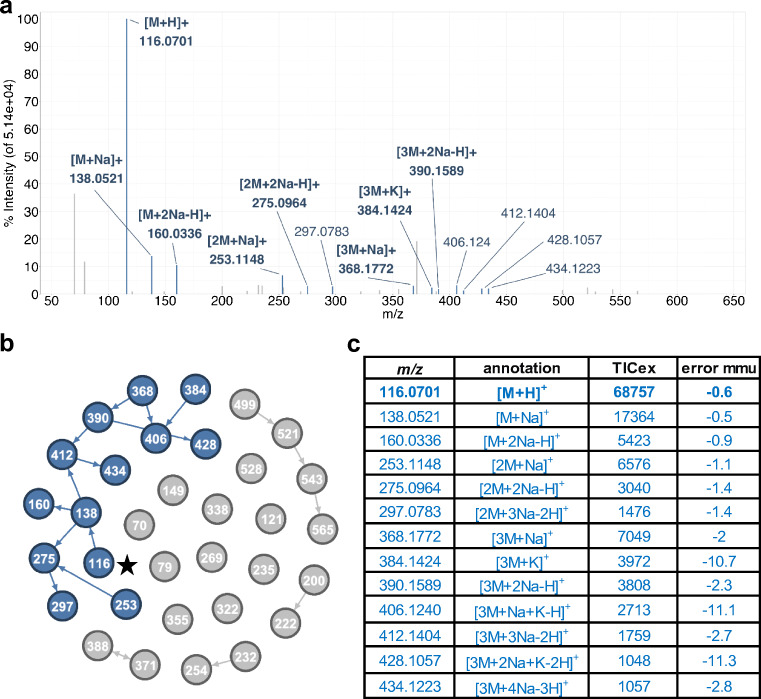
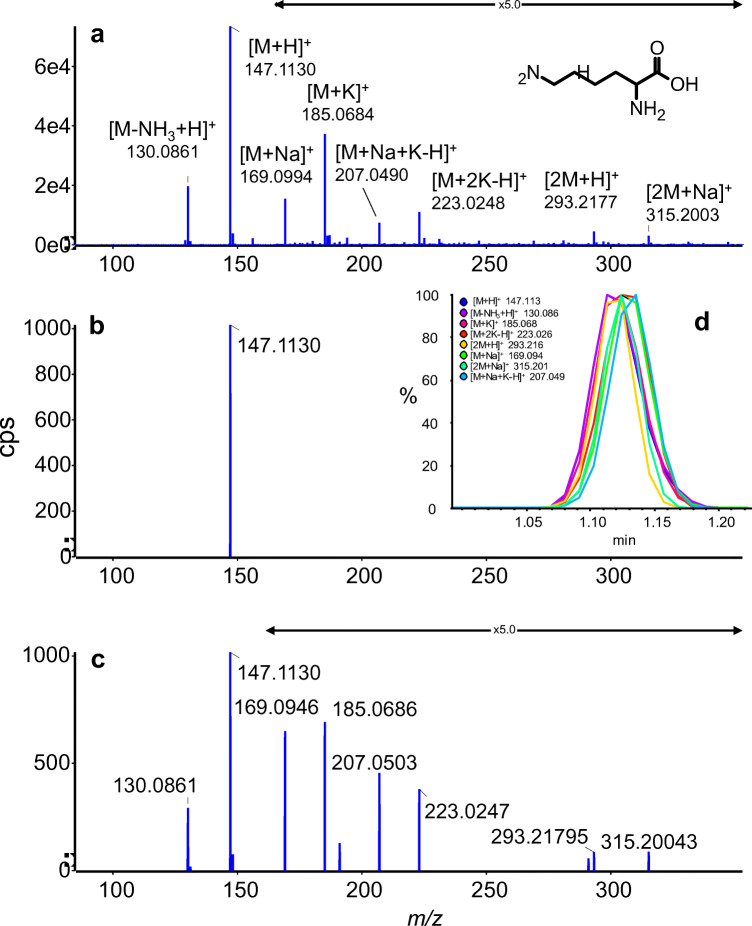
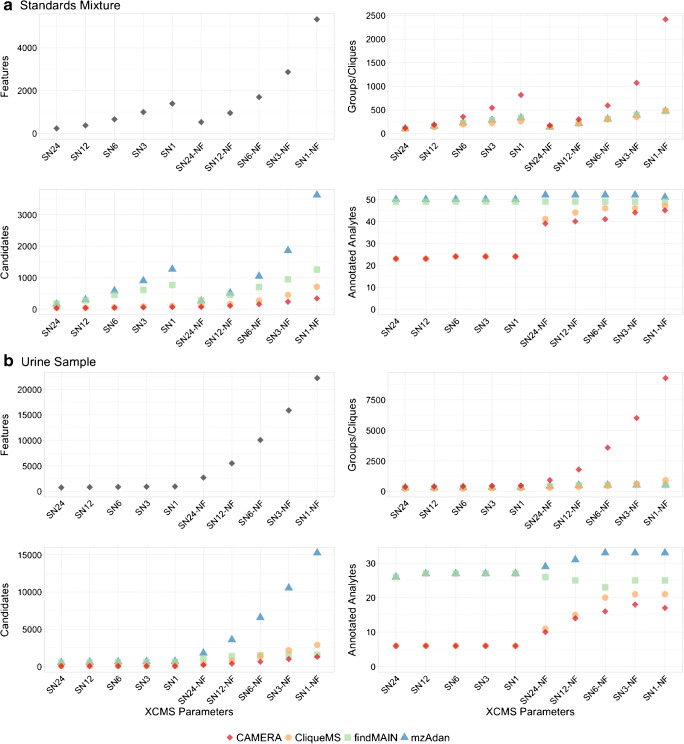
Annotation and interpretation of full scan electrospray mass spectra of metabolites is complicated by the presence of a wide variety of ions. Not only protonated, deprotonated, and neutral loss ions but also sodium, potassium, and ammonium adducts as well as oligomers are frequently observed. This diversity challenges automatic annotation and is often poorly addressed by current annotation tools. In many cases, annotation is integrated in metabolomics workflows and is based on specific chromatographic peak-picking tools. We introduce mzAdan, a nonchromatography-based multipurpose standalone application that was developed for the annotation and exploration of convolved high-resolution ESI-MS spectra. The tool annotates single or multiple accurate mass spectra using a customizable adduct annotation list and outputs a list of [M+H] candidates. MzAdan was first tested with a collection of 408 analytes acquired with flow injection analysis. This resulted in 402 correct [M+H] identifications and, with combinations of sodium, ammonium, and potassium adducts and water and ammonia losses within a tolerance of 10 mmu, explained close to 50% of the total ion current. False positives were monitored with mass accuracy and bias as well as chromatographic behavior which led to the identification of adducts with calcium instead of the expected potassium. MzAdan was then integrated in a workflow with XCMS for the untargeted LC-MS data analysis of a 52 metabolite standard mix and a human urine sample. The results were benchmarked against three other annotation tools, CAMERA, findMAIN, and CliqueMS: findMAIN and mzAdan consistently produced higher numbers of [M+H] candidates compared with CliqueMS and CAMERA, especially with co-eluting metabolites. Detection of low-intensity ions and correct grouping were found to be essential for annotation performance. Graphical abstract.
注释和解释代谢物全扫描电喷雾质谱的复杂性在于存在各种各样的离子。不仅观察到质子化、去质子化和中性丢失离子,还观察到钠、钾和铵加合物以及低聚物。这种多样性对自动注释提出了挑战,目前的注释工具往往无法很好地解决这个问题。在许多情况下,注释是在代谢组学工作流程中进行的,并且基于特定的色谱峰选择工具。我们引入了 mzAdan,这是一种非基于色谱的多用途独立应用程序,用于注释和探索卷积高分辨率 ESI-MS 谱。该工具使用可定制的加合物注释列表注释单个或多个精确质量谱,并输出 [M+H]+候选列表。MzAdan 首先使用流动注射分析获得的 408 种分析物的集合进行了测试。这导致了 402 个正确的 [M+H]+鉴定,并且在 10mmu 的公差内结合了钠、铵和钾加合物以及水和氨的损失,解释了近 50%的总离子流。通过质量精度和偏差以及色谱行为监测假阳性,从而鉴定了预期为钾的钙加合物。MzAdan 然后与 XCMS 集成在一个工作流程中,用于 52 种代谢物标准混合物和人尿液样本的非靶向 LC-MS 数据分析。结果与其他三个注释工具(CAMERA、findMAIN 和 CliqueMS)进行了基准测试:findMAIN 和 mzAdan 与 CliqueMS 和 CAMERA 相比,始终产生更多的 [M+H]+候选物,尤其是对于共洗脱代谢物。低强度离子的检测和正确的分组被发现对注释性能至关重要。