Centre for Heart Lung Innovation, St. Paul's Hospital, 1081 Burrard St., Vancouver, BC, V6Z 1Y6, Canada.
Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada.
Sci Rep. 2020 Nov 4;10(1):19068. doi: 10.1038/s41598-020-76227-7.
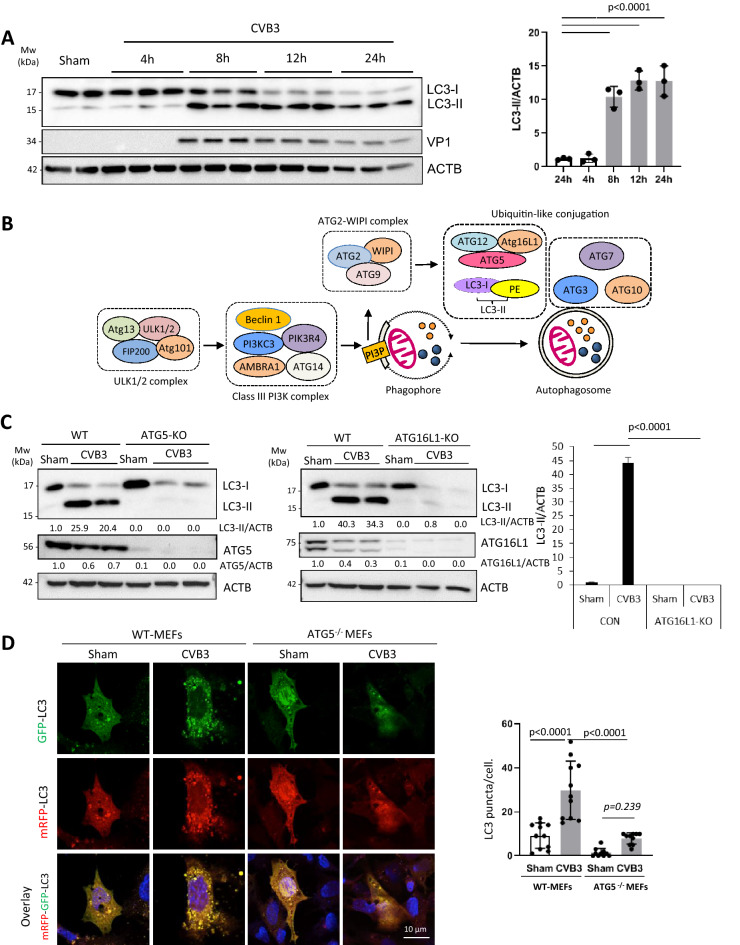
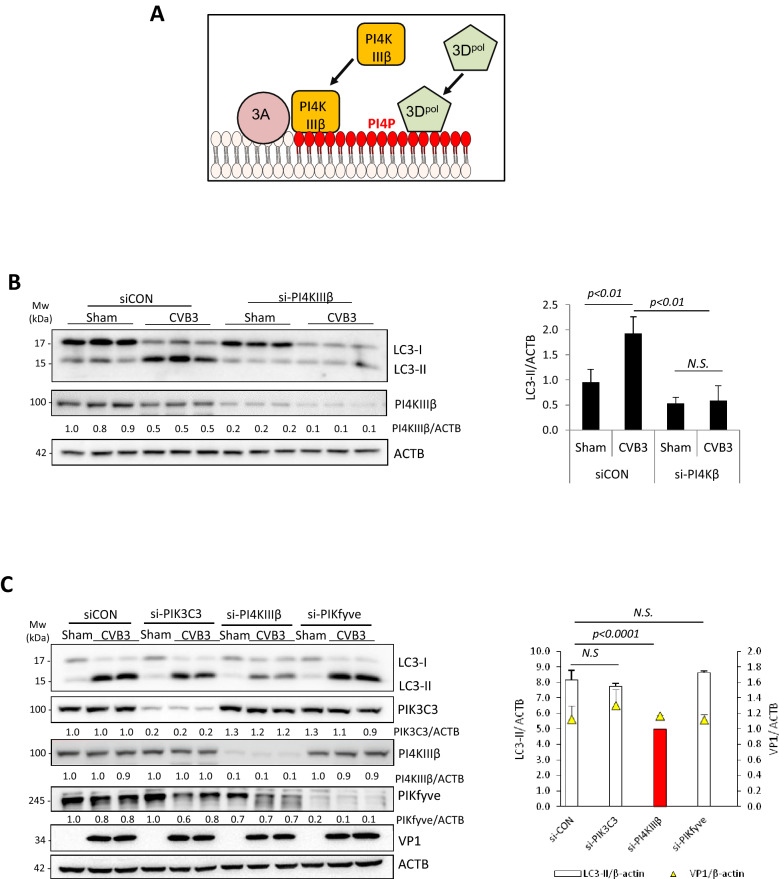
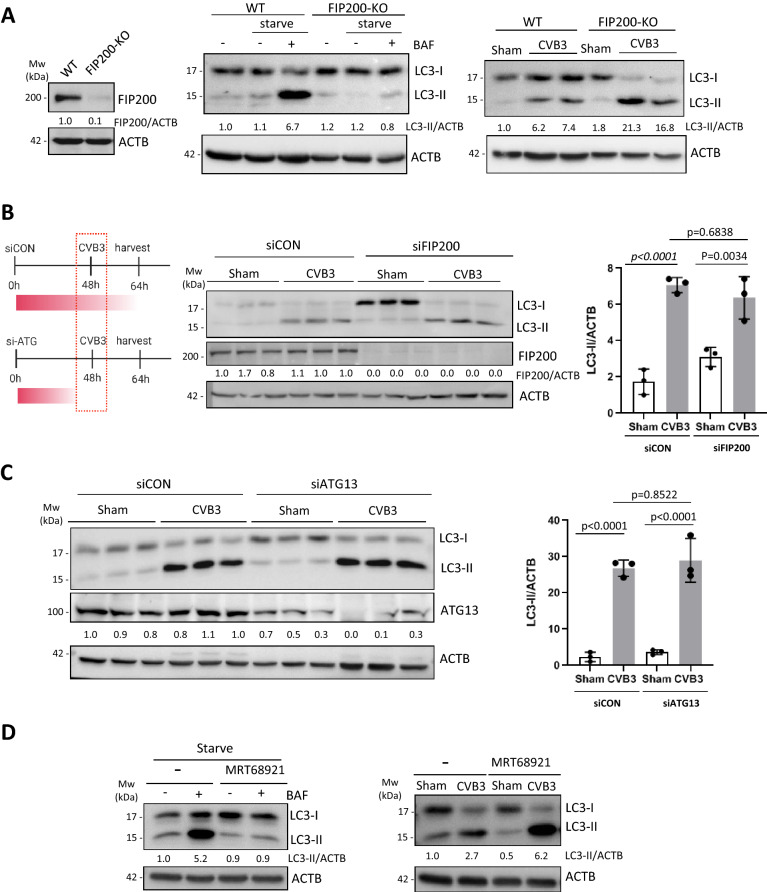
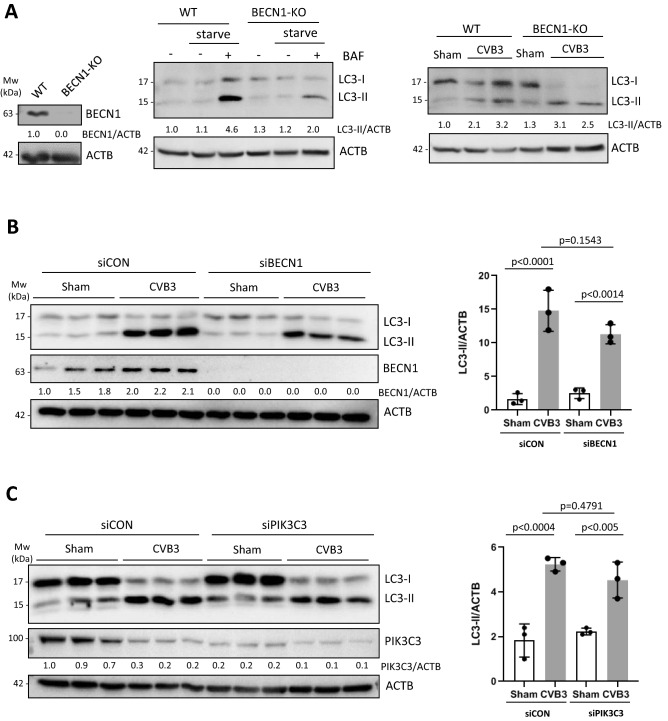
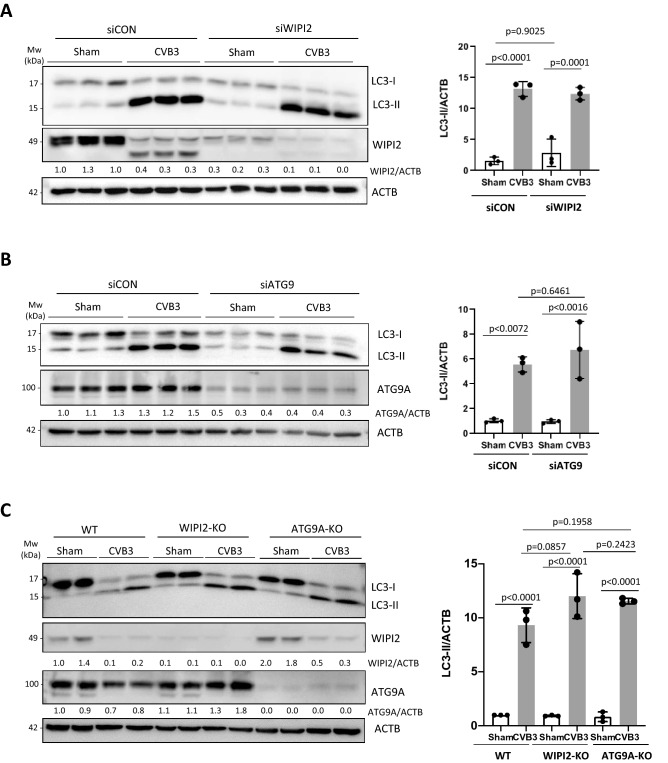
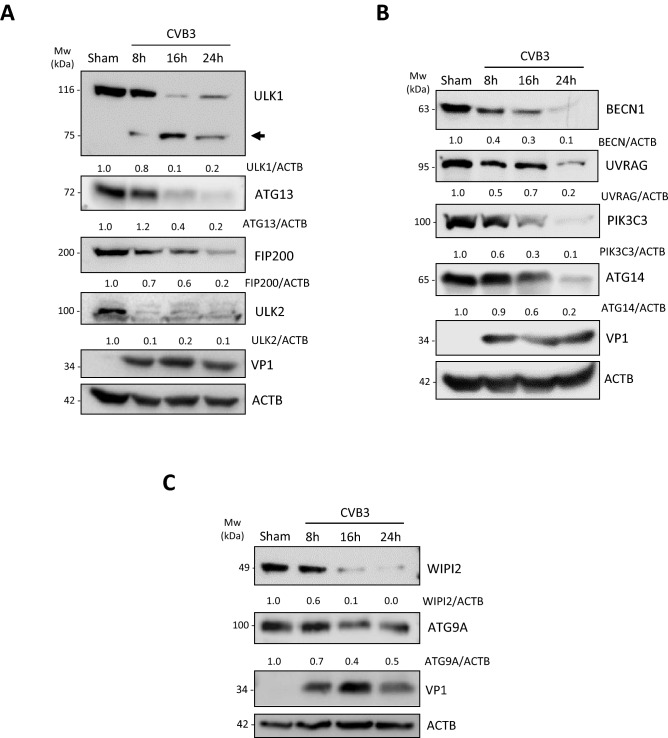
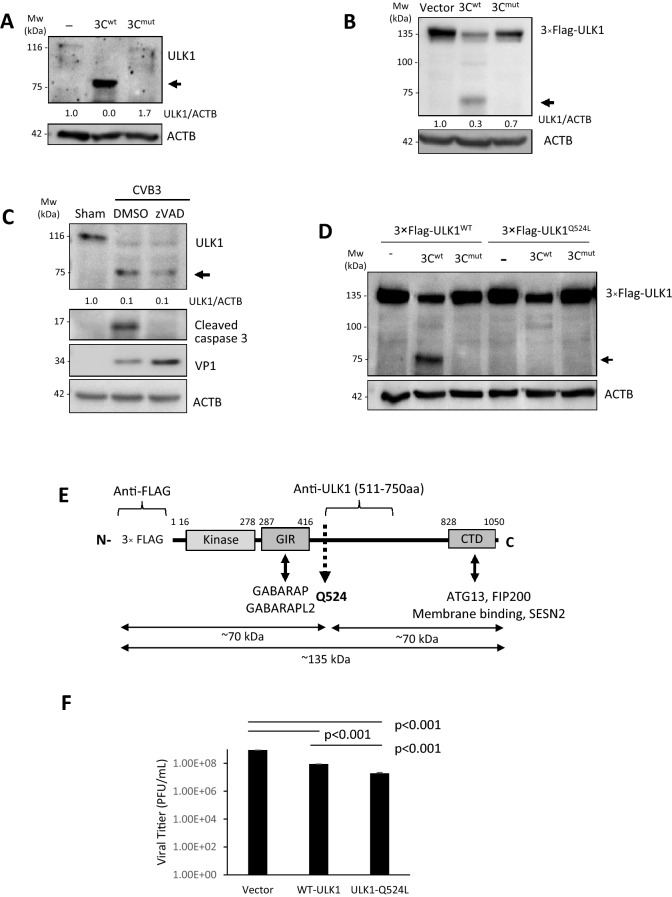
Coxsackievirus B3 (CVB3) is a single-stranded positive RNA virus that usurps cellular machinery, including the evolutionarily anti-viral autophagy pathway, for productive infections. Despite the emergence of double-membraned autophagosome-like vesicles during CVB3 infection, very little is known about the mechanism of autophagy initiation. In this study, we investigated the role of established autophagy factors in the initiation of CVB3-induced autophagy. Using siRNA-mediated gene-silencing and CRISPR-Cas9-based gene-editing in culture cells, we discovered that CVB3 bypasses the ULK1/2 and PI3K complexes to trigger autophagy. Moreover, we found that CVB3-induced LC3 lipidation occurred independent of WIPI2 and the transmembrane protein ATG9 but required components of the late-stage ubiquitin-like ATG conjugation system including ATG5 and ATG16L1. Remarkably, we showed the canonical autophagy factor ULK1 was cleaved through the catalytic activity of the viral proteinase 3C. Mutagenesis experiments identified the cleavage site of ULK1 after Q524, which separates its N-terminal kinase domain from C-terminal substrate binding domain. Finally, we uncovered PI4KIIIβ (a PI4P kinase), but not PI3P or PI5P kinases as requisites for CVB3-induced LC3 lipidation. Taken together, our studies reveal that CVB3 initiates a non-canonical form of autophagy that bypasses ULK1/2 and PI3K signaling pathways to ultimately converge on PI4KIIIβ- and ATG5-ATG12-ATG16L1 machinery.
柯萨奇病毒 B3(CVB3)是一种单链正链 RNA 病毒,它会劫持细胞机制,包括进化上抗病毒的自噬途径,以进行有效的感染。尽管在 CVB3 感染过程中会出现双层类似自噬体的小泡,但对自噬起始的机制知之甚少。在本研究中,我们研究了已建立的自噬因子在 CVB3 诱导的自噬起始中的作用。在培养细胞中使用 siRNA 介导的基因沉默和基于 CRISPR-Cas9 的基因编辑,我们发现 CVB3 绕过 ULK1/2 和 PI3K 复合物来触发自噬。此外,我们发现 CVB3 诱导的 LC3 脂质化发生不依赖于 WIPI2 和跨膜蛋白 ATG9,但需要包括 ATG5 和 ATG16L1 在内的晚期泛素样 ATG 连接系统的成分。值得注意的是,我们表明经典自噬因子 ULK1 通过病毒蛋白酶 3C 的催化活性被切割。突变实验确定了 ULK1 在 Q524 之后的切割位点,该位点将其 N 端激酶结构域与 C 端底物结合结构域分开。最后,我们发现 PI4KIIIβ(一种 PI4P 激酶)而不是 PI3P 或 PI5P 激酶是 CVB3 诱导的 LC3 脂质化所必需的。总之,我们的研究表明,CVB3 启动了一种非典型的自噬形式,绕过 ULK1/2 和 PI3K 信号通路,最终集中在 PI4KIIIβ 和 ATG5-ATG12-ATG16L1 机制上。