Department of Microbiology, National Veterinary Research Institute, Puławy, Poland.
National Food Institute, WHO Collaborating Centre for Antimicrobial Resistance in Foodborne Pathogens, Food and Agriculture Organization Reference Laboratory for Antimicrobial Resistance, and European Union Reference Laboratory for Antimicrobial Resistance, Technical University of Denmark, Kgs. Lyngby, Denmark.
PLoS One. 2020 Dec 3;15(12):e0242987. doi: 10.1371/journal.pone.0242987. eCollection 2020.
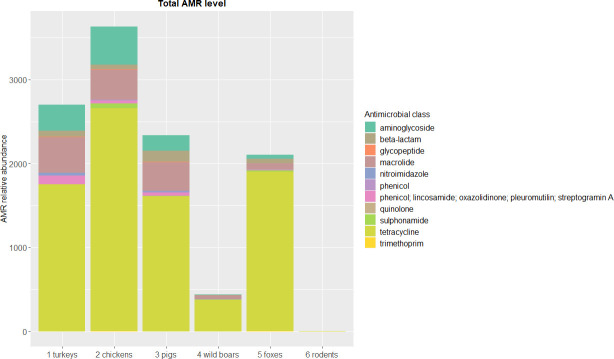
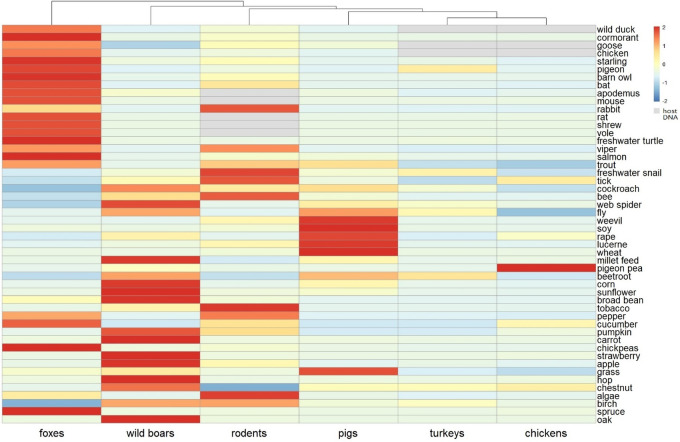
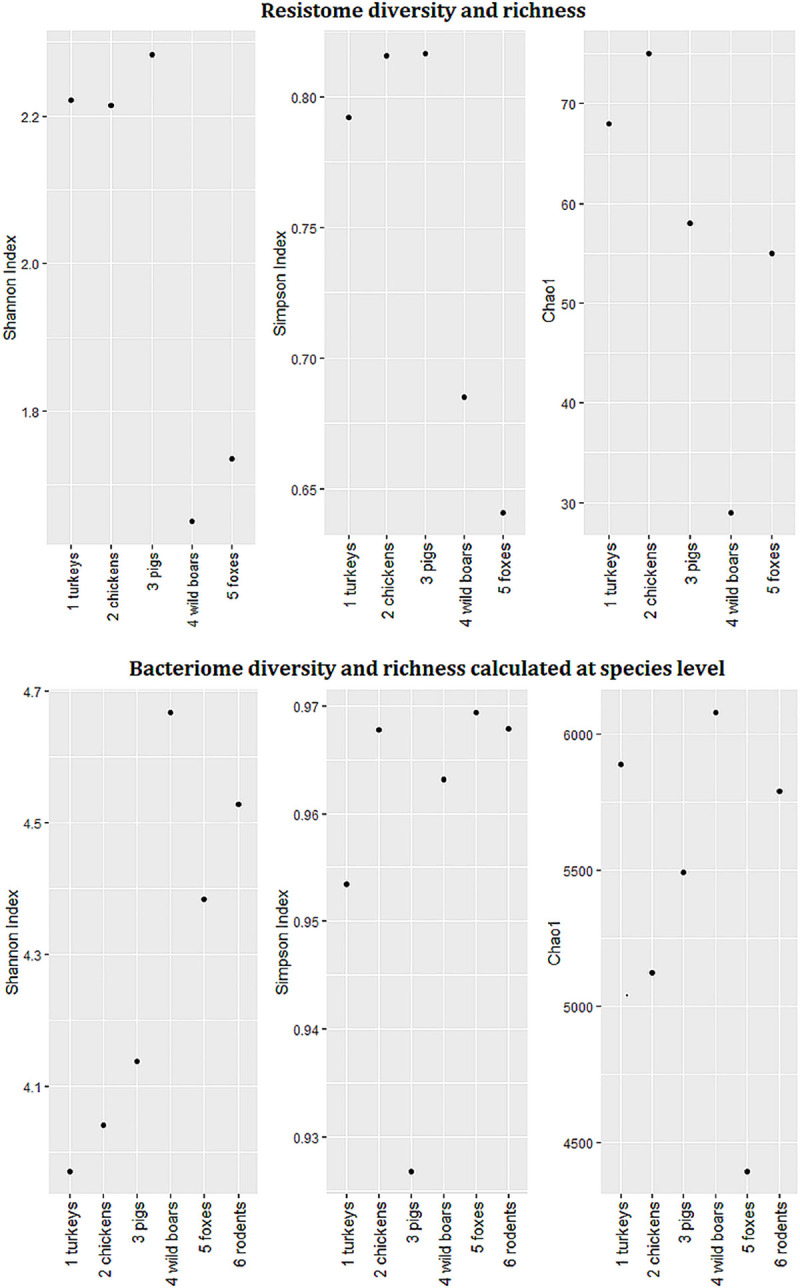
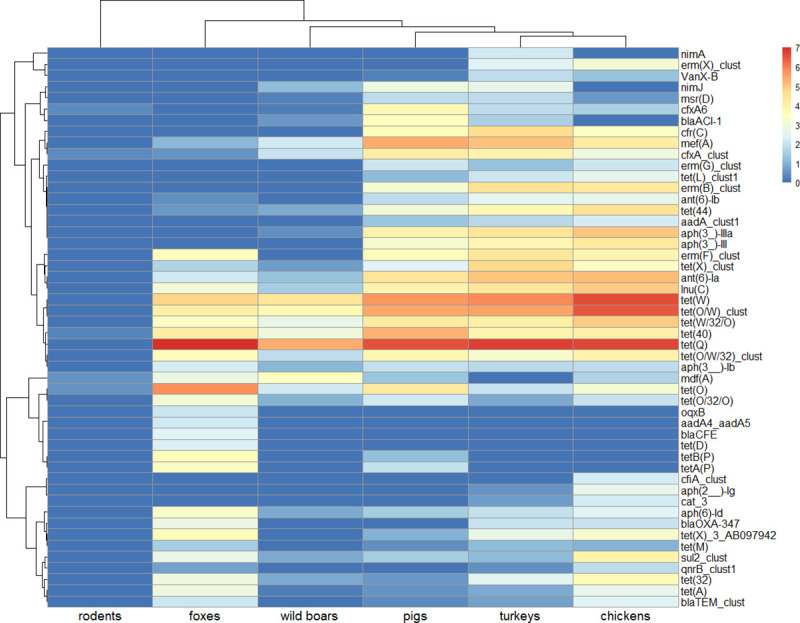
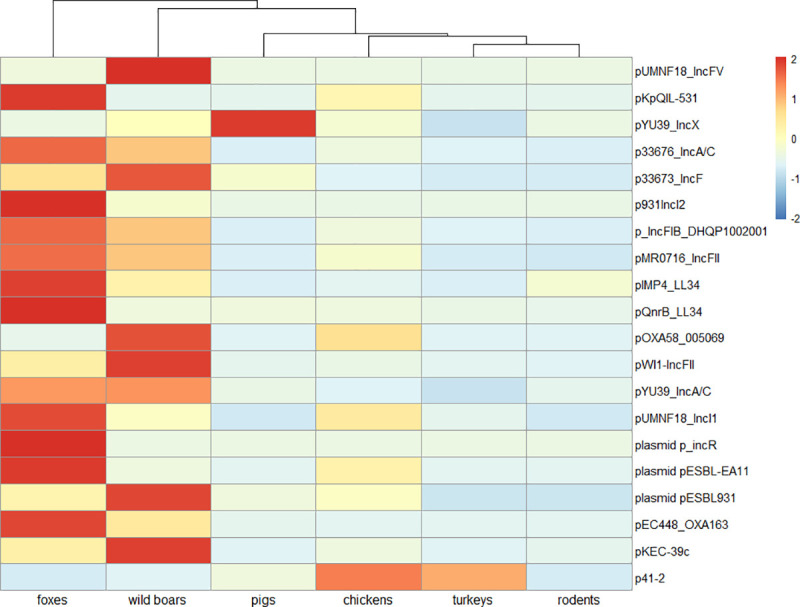
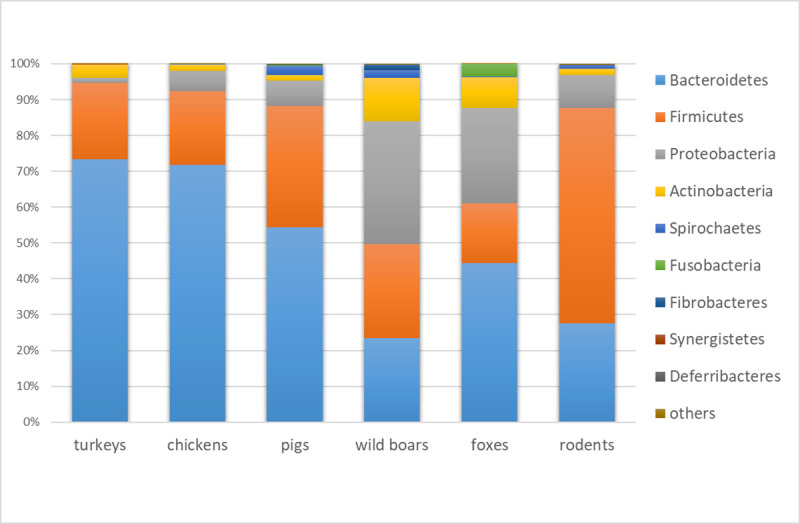
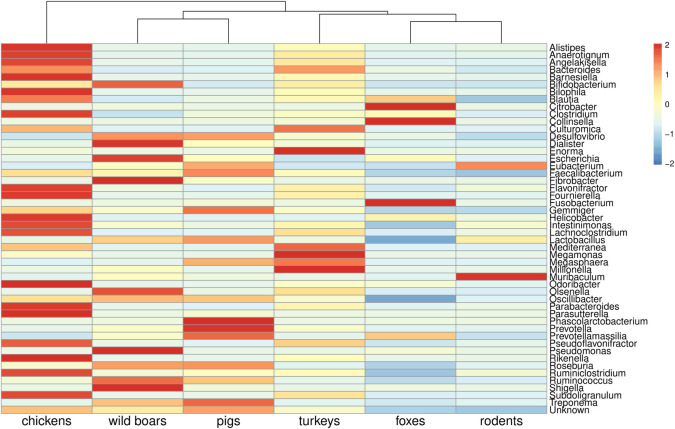
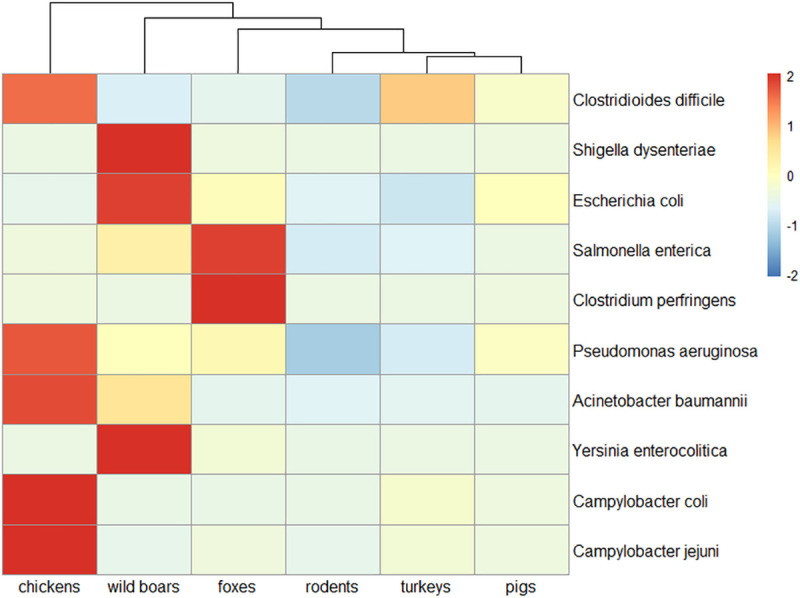
Antimicrobial resistance (AMR) in bacteria is a complex subject, why one need to look at this phenomenon from a wider and holistic perspective. The extensive use of the same antimicrobial classes in human and veterinary medicine as well as horticulture is one of the main drivers for the AMR selection. Here, we applied shotgun metagenomics to investigate the AMR epidemiology in several animal species including farm animals, which are often exposed to antimicrobial treatment opposed to an unique set of wild animals that seems not to be subjected to antimicrobial pressure. The comparison of the domestic and wild animals allowed to investigate the possible anthropogenic impact on AMR spread. Inclusion of animals with different feeding behaviors (carnivores, omnivores) enabled to further assess which AMR genes that thrives within the food chain. We tested fecal samples not only of intensively produced chickens, turkeys, and pigs, but also of wild animals such as wild boars, red foxes, and rodents. A multi-directional approach mapping obtained sequences to several databases provided insight into the occurrence of the different AMR genes. The method applied enabled also analysis of other factors that may influence AMR of intestinal microbiome such as diet. Our findings confirmed higher levels of AMR in farm animals than in wildlife. The results also revealed the potential of wildlife in the AMR dissemination. Particularly in red foxes, we found evidence of several AMR genes conferring resistance to critically important antimicrobials like quinolones and cephalosporins. In contrast, the lowest abundance of AMR was observed in rodents originating from natural environment with presumed limited exposure to antimicrobials. Shotgun metagenomics enabled us to demonstrate that discrepancies between AMR profiles found in the intestinal microbiome of various animals probably resulted from the different antimicrobial exposure, habitats, and behavior of the tested animal species.
细菌的抗微生物药物耐药性(AMR)是一个复杂的问题,因此需要从更广泛和整体的角度来看待这一现象。在人类和兽医医学以及园艺中广泛使用相同的抗微生物药物类别是 AMR 选择的主要驱动因素之一。在这里,我们应用鸟枪法宏基因组学来研究包括农场动物在内的几种动物物种中的 AMR 流行病学,这些动物经常接触抗微生物治疗,而不是一组独特的野生动物,它们似乎不受抗微生物压力的影响。对家养动物和野生动物的比较可以研究人为因素对抗微生物药物耐药性传播的可能影响。纳入具有不同饲养行为(肉食动物、杂食动物)的动物可以进一步评估哪些抗微生物药物基因在食物链中占优势。我们不仅测试了集约化生产的鸡、火鸡和猪的粪便样本,还测试了野猪、赤狐和啮齿动物等野生动物的粪便样本。将获得的序列映射到多个数据库的多方向方法提供了对抗微生物药物耐药性基因的发生情况的深入了解。应用的方法还允许分析可能影响肠道微生物组抗微生物药物耐药性的其他因素,如饮食。我们的研究结果证实了农场动物的抗微生物药物耐药性水平高于野生动物。研究结果还揭示了野生动物在抗微生物药物耐药性传播中的潜力。特别是在赤狐中,我们发现了一些抗微生物药物耐药基因的证据,这些基因对抗菌素如喹诺酮类和头孢菌素类具有耐药性。相比之下,在来自自然环境、假定接触抗微生物药物有限的啮齿动物中,抗微生物药物耐药性的丰度最低。鸟枪法宏基因组学使我们能够证明,不同动物肠道微生物组中发现的抗微生物药物耐药性谱之间的差异可能是由于不同的抗微生物药物暴露、栖息地和测试动物物种的行为所致。