Goodall Rhys E A, Lee Alpha A
University of Cambridge, Cavendish Laboratory, Cambridge, UK.
Nat Commun. 2020 Dec 8;11(1):6280. doi: 10.1038/s41467-020-19964-7.
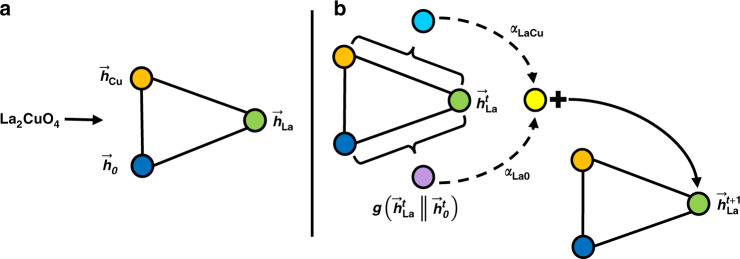
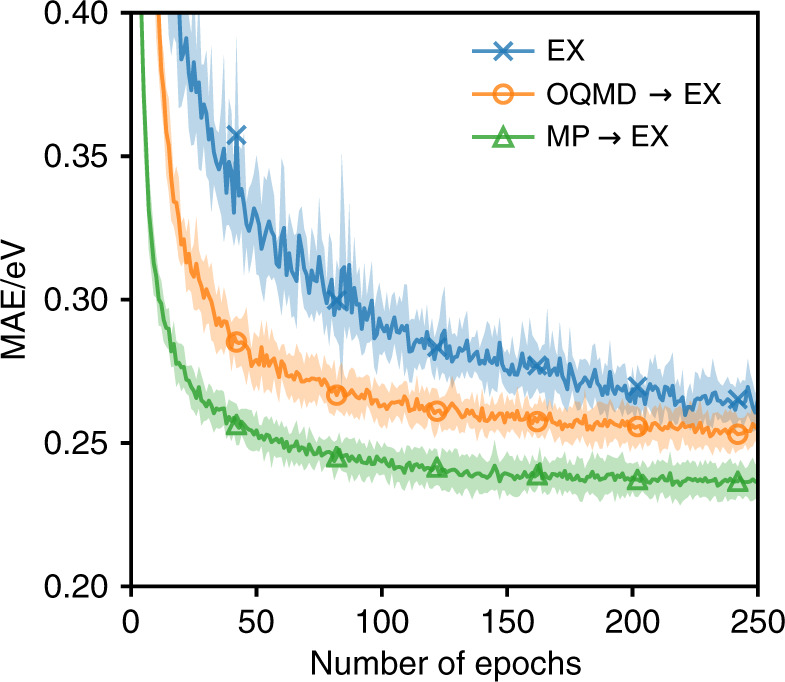
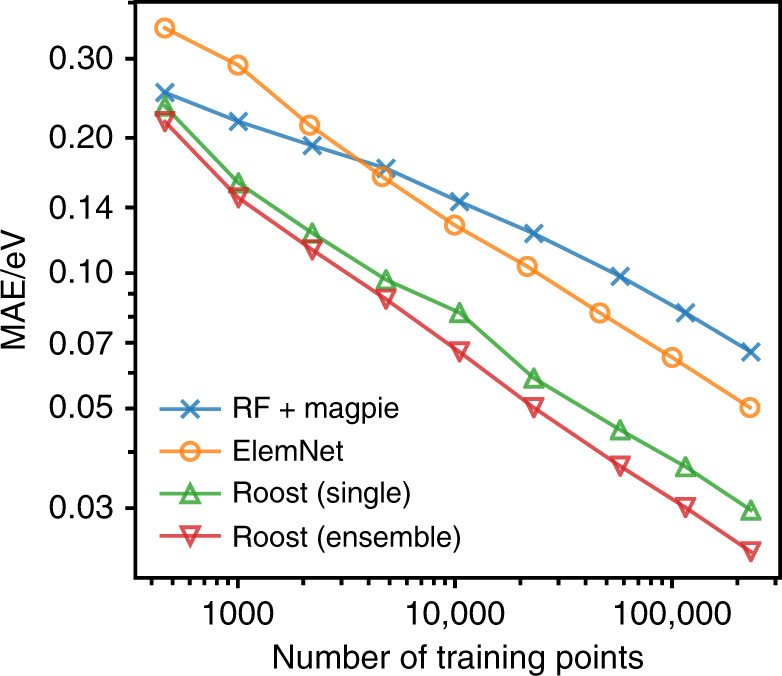
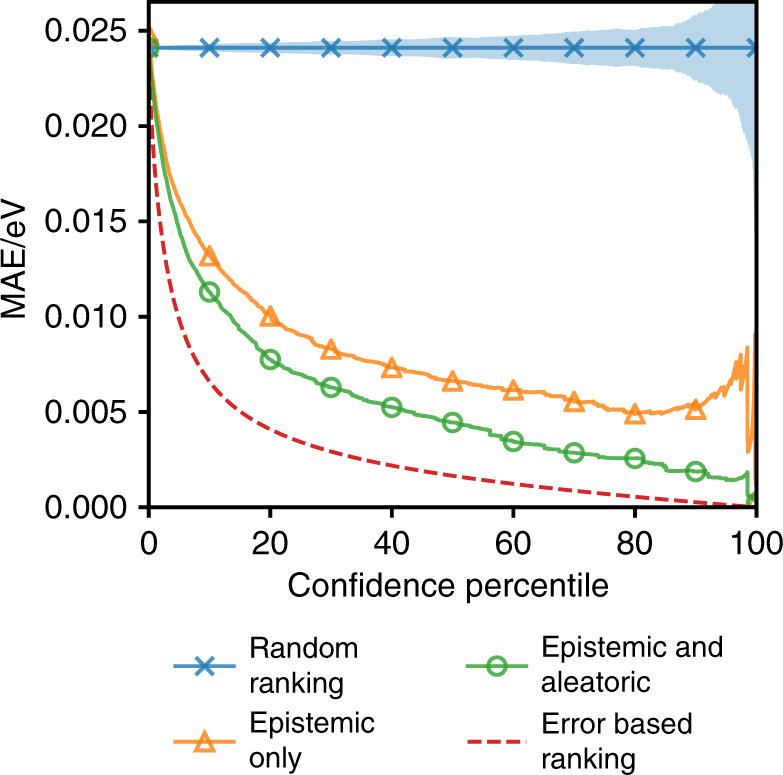
Machine learning has the potential to accelerate materials discovery by accurately predicting materials properties at a low computational cost. However, the model inputs remain a key stumbling block. Current methods typically use descriptors constructed from knowledge of either the full crystal structure - therefore only applicable to materials with already characterised structures - or structure-agnostic fixed-length representations hand-engineered from the stoichiometry. We develop a machine learning approach that takes only the stoichiometry as input and automatically learns appropriate and systematically improvable descriptors from data. Our key insight is to treat the stoichiometric formula as a dense weighted graph between elements. Compared to the state of the art for structure-agnostic methods, our approach achieves lower errors with less data.
机器学习有潜力通过以低计算成本准确预测材料特性来加速材料发现。然而,模型输入仍然是一个关键的绊脚石。当前方法通常使用从完整晶体结构知识构建的描述符——因此仅适用于具有已表征结构的材料——或从化学计量学手工设计的与结构无关的固定长度表示。我们开发了一种机器学习方法,该方法仅将化学计量学作为输入,并从数据中自动学习合适且可系统改进的描述符。我们的关键见解是将化学计量式视为元素之间的密集加权图。与无结构方法的现有技术相比,我们的方法在数据较少的情况下实现了更低的误差。