Baeza J Antonio
Department of Biological Sciences, Clemson University, 132 Long Hall, Clemson, SC, 29634, USA.
Smithsonian Marine Station at Fort Pierce, 701 Seaway Drive, Fort Pierce, Florida, 34949, USA.
BMC Genomics. 2020 Dec 9;21(1):882. doi: 10.1186/s12864-020-07292-5.
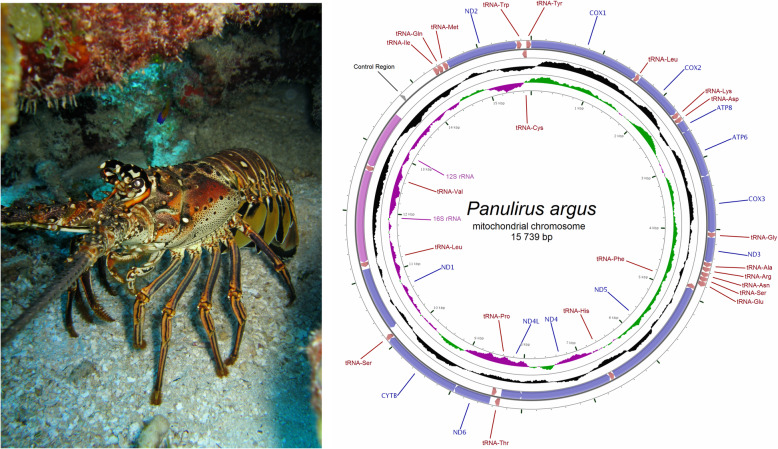
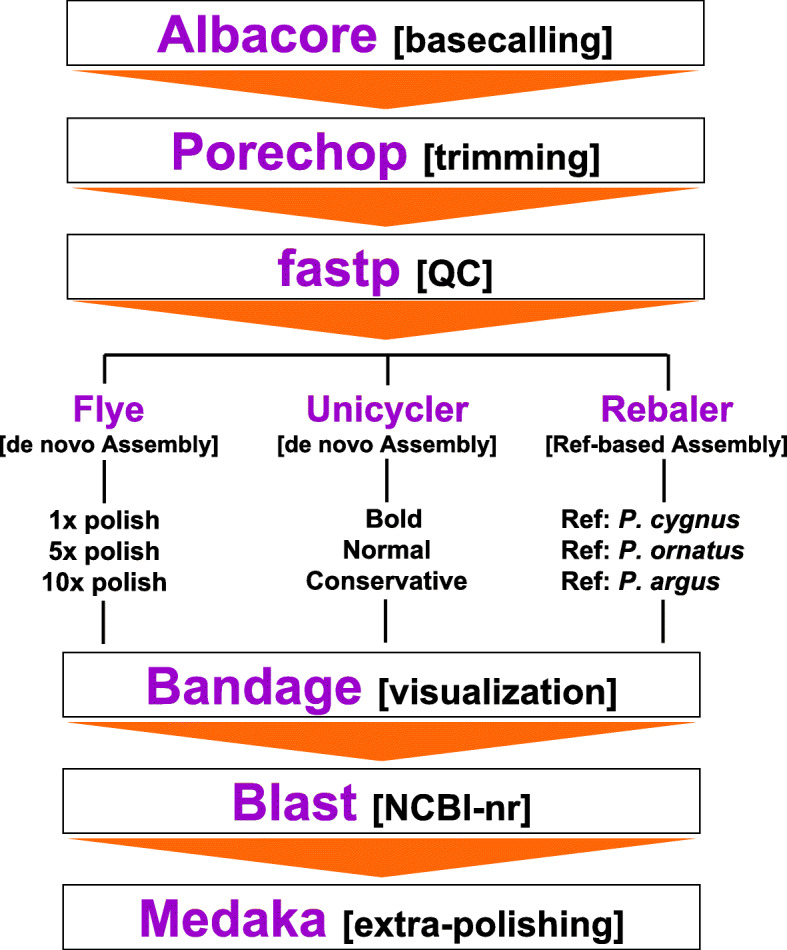
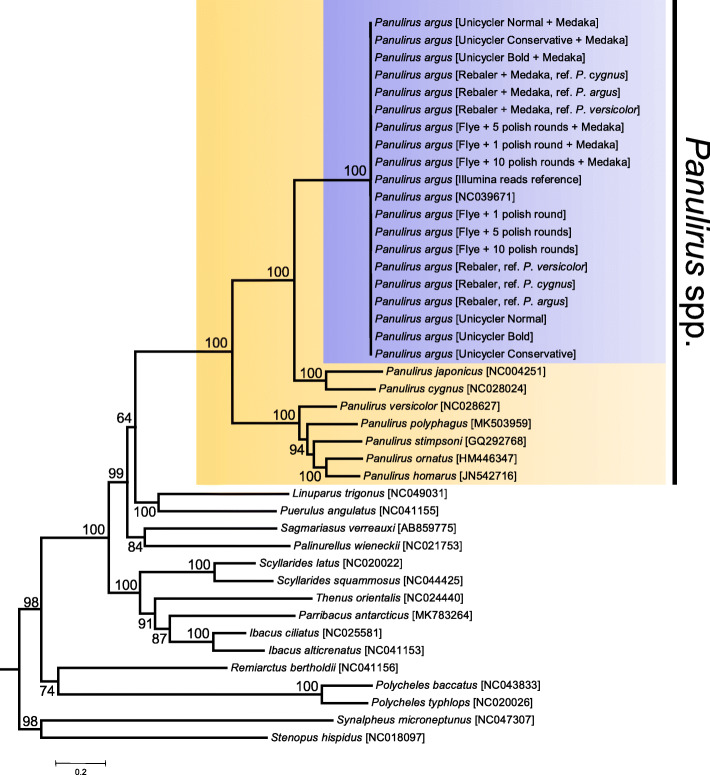
Whole mitogenomes or short fragments (i.e., 300-700 bp of the cox1 gene) are the markers of choice for revealing within- and among-species genealogies. Protocols for sequencing and assembling mitogenomes include 'primer walking' or 'long PCR' followed by Sanger sequencing or Illumina short-read low-coverage whole genome (LC-WGS) sequencing with or without prior enrichment of mitochondrial DNA. The aforementioned strategies assemble complete and accurate mitochondrial genomes but are time consuming and/or expensive. In this study, I first tested whether mitogenomes can be sequenced from long-read nanopore sequencing data exclusively. Second, I explored the accuracy of the long-read assembled genomes by comparing them to a 'gold' standard reference mitogenome retrieved from the same individual using Illumina sequencing. Third and lastly, I tested if the long-read assemblies are useful for mitophylogenomics and barcoding research. To accomplish these goals, I used the Caribbean spiny lobster Panulirus argus, an ecologically relevant species in shallow water coral reefs and target of the most lucrative fishery in the greater Caribbean region.
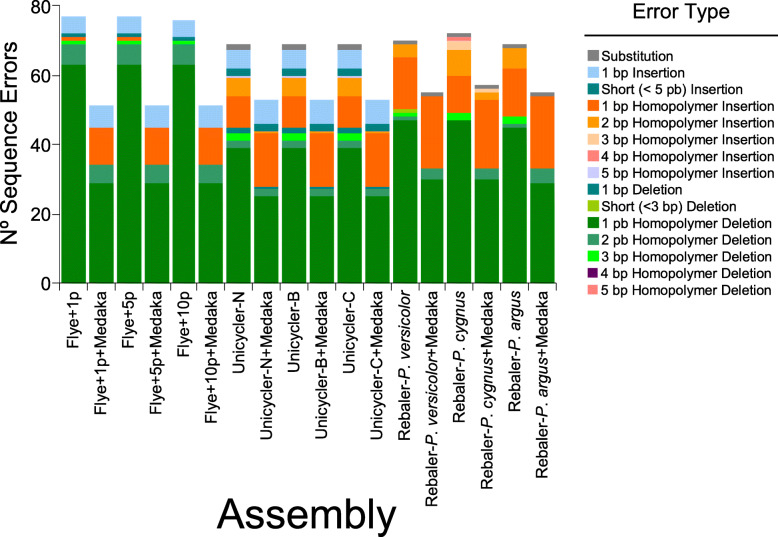
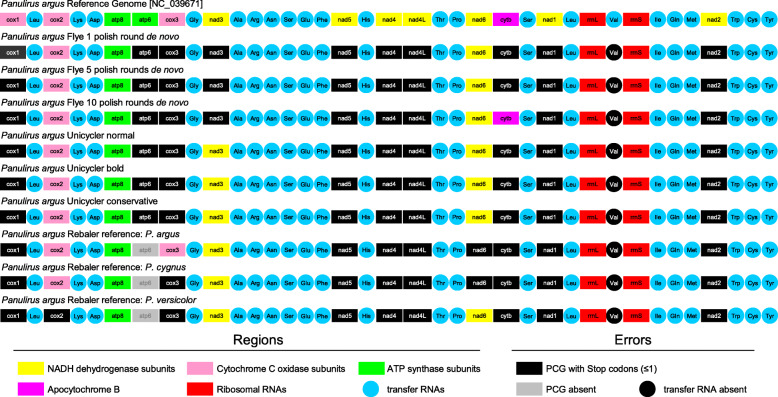
LC-WGS using a MinION ONT device and various de-novo and reference-based assembly pipelines retrieved a complete and highly accurate mitogenome for the Caribbean spiny lobster Panulirus argus. Discordance between each of the long-read assemblies and the reference mitogenome was mostly due to indels at the flanks of homopolymer regions. Although not 'perfect', phylogenetic analyses using entire mitogenomes or a fragment of the cox1 gene demonstrated that mitogenomes assembled using long reads reliably identify the sequenced specimen as belonging to P. argus and distinguish it from other related species in the same genus, family, and superorder.
This study serves as a proof-of-concept for the future implementation of in-situ surveillance protocols using the MinION to detect mislabeling in P. argus across its supply chain. Mislabeling detection will improve fishery management in this overexploited lobster. This study will additionally aid in decreasing costs for exploring meta-population connectivity in the Caribbean spiny lobster and will aid with the transfer of genomics technology to low-income countries.
完整的线粒体基因组或短片段(即细胞色素氧化酶亚基1基因的300 - 700bp)是揭示物种内和物种间谱系的首选标记。线粒体基因组测序和组装方案包括“引物步移法”或“长片段PCR”,随后进行桑格测序或Illumina短读长低覆盖度全基因组(LC - WGS)测序,测序前可对线粒体DNA进行富集或不富集。上述策略能够组装出完整且准确的线粒体基因组,但耗时且/或成本高昂。在本研究中,我首先测试了是否仅通过长读长纳米孔测序数据就能对线粒体基因组进行测序。其次,我通过将长读长组装的基因组与使用Illumina测序从同一个体获取的“黄金”标准参考线粒体基因组进行比较,探究了长读长组装基因组的准确性。第三也是最后一点,我测试了长读长组装是否对线粒体系统发育基因组学和条形码研究有用。为实现这些目标,我使用了加勒比刺龙虾(Panulirus argus),这是一种在浅水珊瑚礁中具有生态意义的物种,也是大加勒比地区最具商业价值渔业的捕捞对象。
使用MinION ONT设备进行LC - WGS,并采用各种从头组装和基于参考的组装流程,为加勒比刺龙虾(Panulirus argus)获得了一个完整且高度准确的线粒体基因组。每个长读长组装与参考线粒体基因组之间的不一致主要是由于同聚物区域侧翼的插入缺失。尽管并非“完美”,但使用完整线粒体基因组或细胞色素氧化酶亚基1基因片段进行的系统发育分析表明,使用长读长组装的线粒体基因组能够可靠地将测序样本鉴定为属于Panulirus argus,并将其与同一属、科和总目中的其他相关物种区分开来。
本研究为未来使用MinION实施原位监测方案以检测Panulirus argus整个供应链中的标签错误提供了概念验证。标签错误检测将改善这种过度捕捞龙虾的渔业管理。本研究还将有助于降低探索加勒比刺龙虾种群连通性的成本,并有助于将基因组技术转移到低收入国家。