Key Laboratory of Tropical Translational Medicine of Ministry of Education and School of Tropical Medicine and Laboratory Medicine, Hainan Medical University, Haikou, China.
CAS Center for Excellence in Biotic Interactions, University of Chinese Academy of Sciences, Beijing, China.
Front Cell Infect Microbiol. 2020 Dec 15;10:596750. doi: 10.3389/fcimb.2020.596750. eCollection 2020.
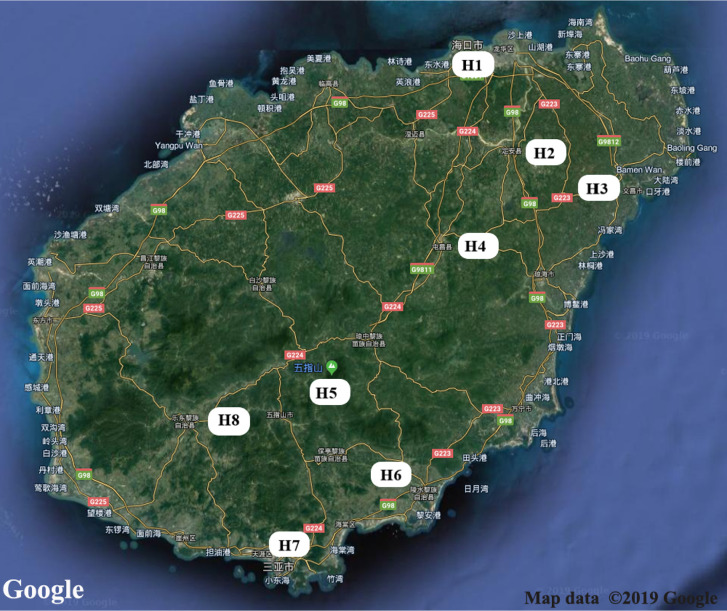
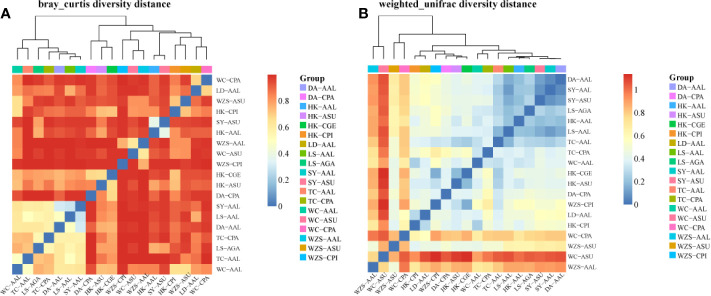
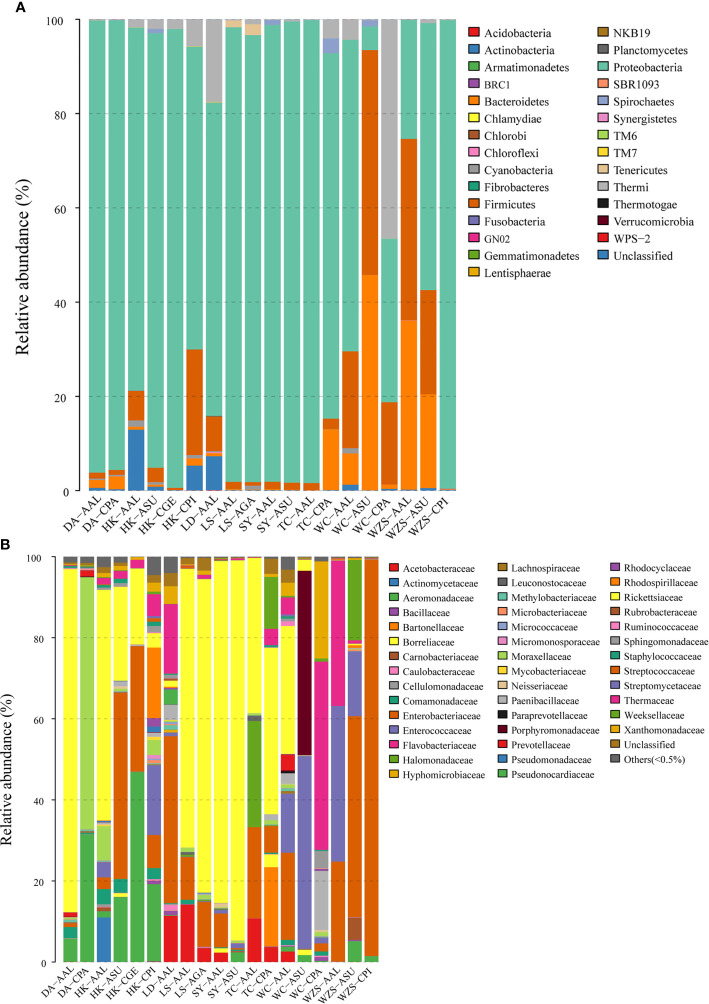
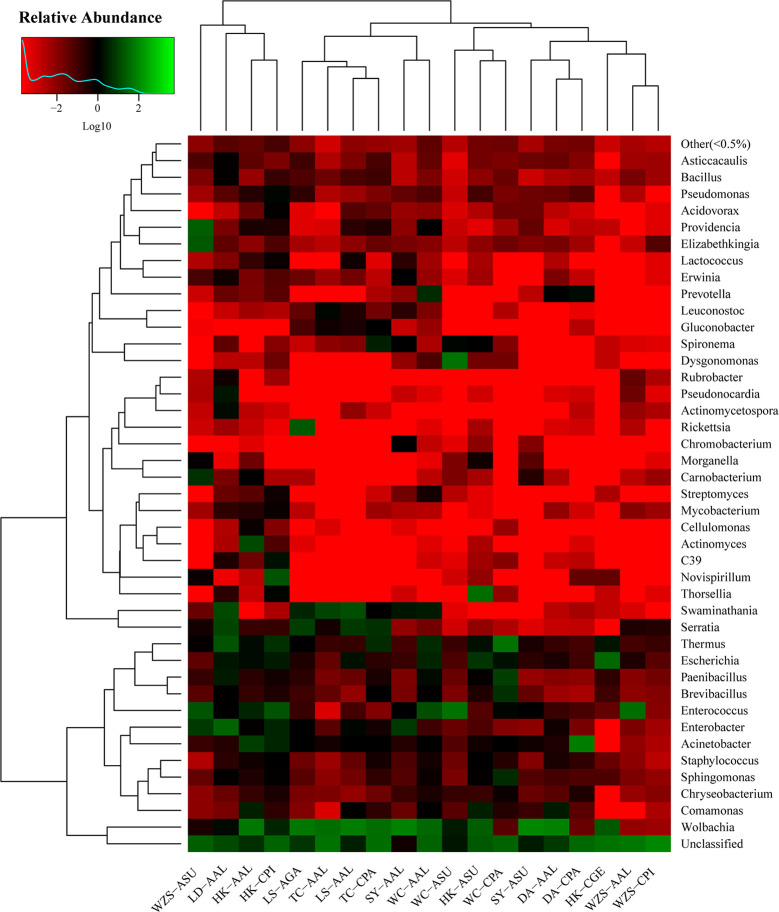
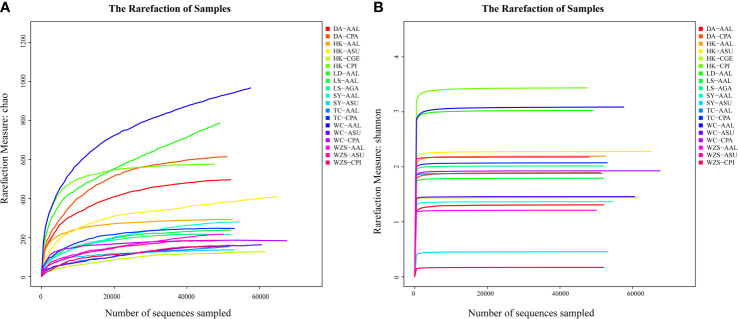
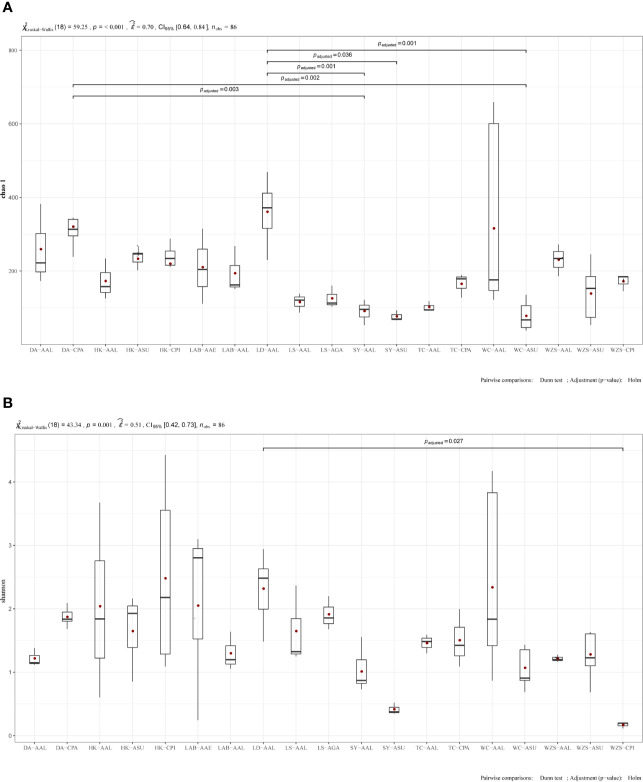
The midgut microbial community composition, structure, and function of field-collected mosquitoes may provide a way to exploit microbial function for mosquito-borne disease control. However, it is unclear how adult mosquitoes acquire their microbiome, how the microbiome affects life history traits and how the microbiome influences community structure. We analyzed the composition of 501 midgut bacterial communities from field-collected adult female mosquitoes, including , , , , and , across eight habitats using the HiSeq 4000 system and the V3-V4 hyper-variable region of 16S rRNA gene. After quality filtering and rarefaction, a total of 1421 operational taxonomic units, belonging to 29 phyla, 44 families, and 43 genera were identified. (75.67%) were the most common phylum, followed by (10.38%), (6.87%), (4.60%), and (1.58%). The genera (33.00%), (20.27%), (7.49%), (7.00%), (4.52%), and (4.31%) were dominant in the samples analyzed and accounted for 76.59% of the total genera. We characterized the midgut bacterial communities of six mosquito species in Hainan province, China. The gut bacterial communities were different in composition and abundance, among locations, for all mosquito species. There were significant differences in the gut microbial composition between some species and substantial variation in the gut microbiota between individuals of the same mosquito species. There was a marked variation in different mosquito gut microbiota within the same location. These results might be useful in the identification of microbial communities that could be exploited for disease control.
从野外采集的成年雌性蚊子的中肠微生物群落组成、结构和功能中,可以了解到微生物功能可能是控制蚊媒疾病的一种方法。然而,目前尚不清楚成年蚊子如何获得其微生物组,微生物组如何影响生活史特征,以及微生物组如何影响群落结构。我们使用 HiSeq 4000 系统和 16S rRNA 基因的 V3-V4 高变区,对来自 8 种生境的 501 个野外采集的成年雌性蚊子的中肠细菌群落的组成进行了分析,包括 、 、 、 和 。经过质量过滤和稀疏处理,共鉴定出 1421 个操作分类单元,属于 29 个门、44 个科和 43 个属。 (75.67%)是最常见的门,其次是 (10.38%)、 (6.87%)、 (4.60%)和 (1.58%)。 (33.00%)、 (20.27%)、 (7.49%)、 (7.00%)、 (4.52%)和 (4.31%)是分析样本中的优势属,占总属的 76.59%。我们对中国海南省的 6 种蚊子的中肠细菌群落进行了特征描述。肠道细菌群落的组成和丰度在不同地点的蚊子种类之间存在差异。一些物种的肠道微生物组成存在显著差异,同一蚊种个体之间的肠道微生物也存在很大差异。同一地点不同蚊子的肠道微生物群有明显的变化。这些结果可能有助于识别可用于疾病控制的微生物群落。