Division of Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands.
UMR 518 MIA-Paris, INRAE, c/o 113 rue Nationale, 75103, Paris, France.
Sci Rep. 2021 Jan 13;11(1):947. doi: 10.1038/s41598-020-80363-5.
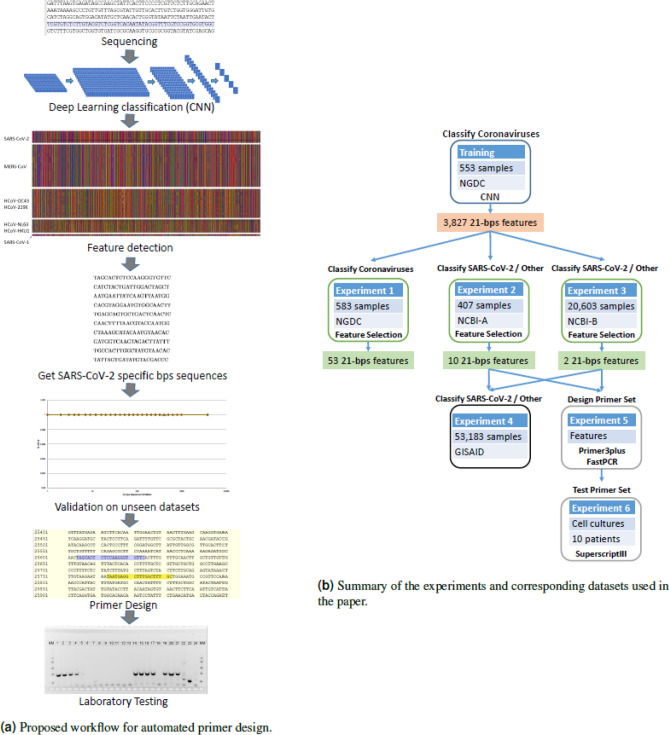
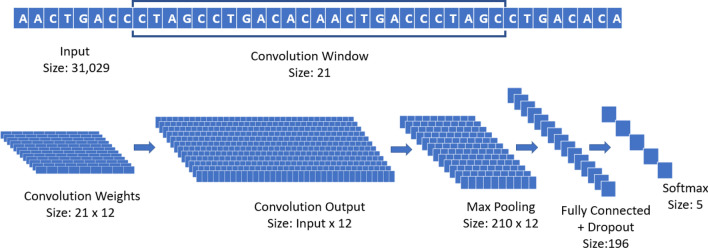
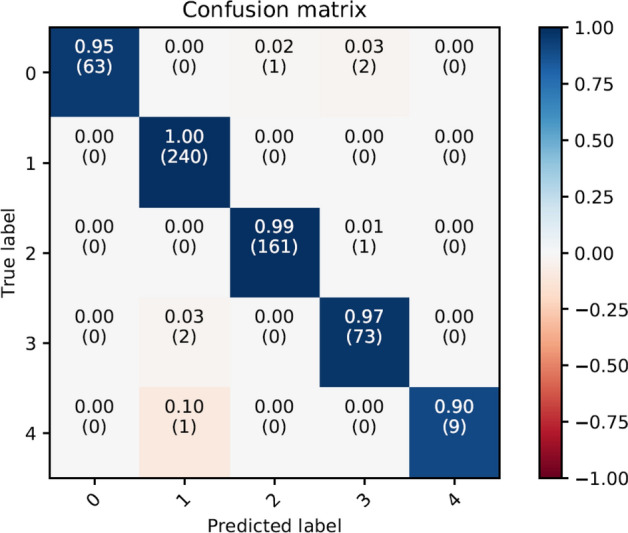
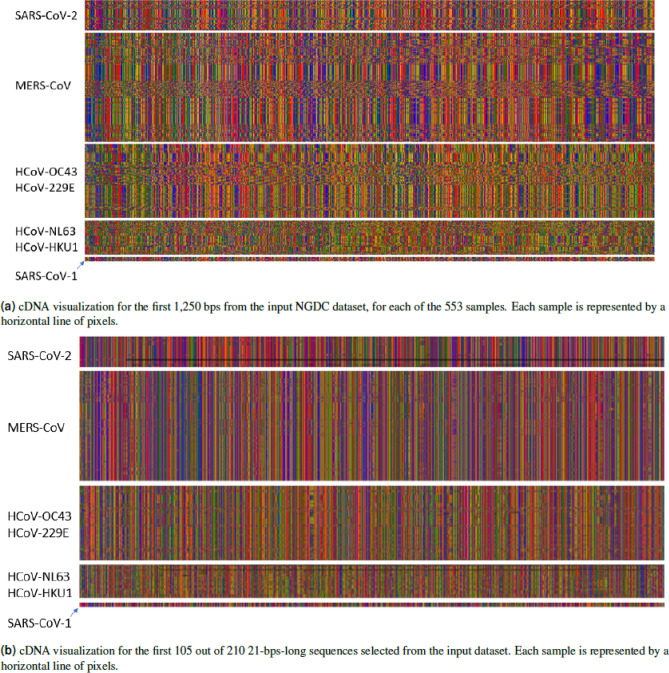
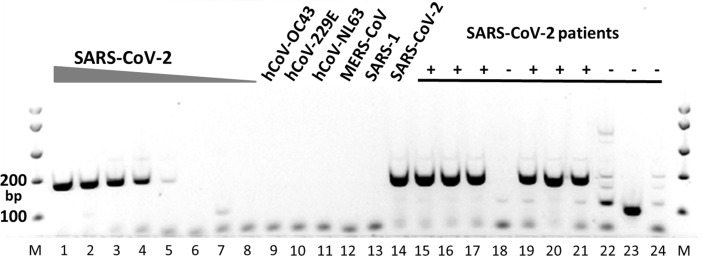
In this paper, deep learning is coupled with explainable artificial intelligence techniques for the discovery of representative genomic sequences in SARS-CoV-2. A convolutional neural network classifier is first trained on 553 sequences from the National Genomics Data Center repository, separating the genome of different virus strains from the Coronavirus family with 98.73% accuracy. The network's behavior is then analyzed, to discover sequences used by the model to identify SARS-CoV-2, ultimately uncovering sequences exclusive to it. The discovered sequences are validated on samples from the National Center for Biotechnology Information and Global Initiative on Sharing All Influenza Data repositories, and are proven to be able to separate SARS-CoV-2 from different virus strains with near-perfect accuracy. Next, one of the sequences is selected to generate a primer set, and tested against other state-of-the-art primer sets, obtaining competitive results. Finally, the primer is synthesized and tested on patient samples (n = 6 previously tested positive), delivering a sensitivity similar to routine diagnostic methods, and 100% specificity. The proposed methodology has a substantial added value over existing methods, as it is able to both automatically identify promising primer sets for a virus from a limited amount of data, and deliver effective results in a minimal amount of time. Considering the possibility of future pandemics, these characteristics are invaluable to promptly create specific detection methods for diagnostics.
在本文中,深度学习与可解释人工智能技术相结合,用于发现 SARS-CoV-2 中有代表性的基因组序列。首先,在国家基因组学数据中心存储库的 553 个序列上训练卷积神经网络分类器,该分类器以 98.73%的准确率将不同病毒株的基因组与冠状病毒家族区分开来。然后,分析网络的行为,以发现模型用于识别 SARS-CoV-2 的序列,最终揭示其独有的序列。在所发现的序列上,对来自国家生物技术信息中心和全球共享所有流感数据倡议存储库的样本进行验证,结果表明,它们能够以近乎完美的准确度将 SARS-CoV-2 与不同的病毒株区分开来。接下来,选择其中一条序列生成一组引物,并与其他最先进的引物组进行测试,结果具有竞争力。最后,合成引物并在患者样本(n=6 个先前检测呈阳性的样本)上进行测试,结果与常规诊断方法具有相似的灵敏度,且特异性为 100%。与现有方法相比,该方法具有实质性的附加价值,因为它不仅能够从有限的数据中自动识别病毒的有前途的引物组,而且能够在最短的时间内提供有效的结果。考虑到未来可能发生大流行的情况,这些特征对于及时创建特定的诊断检测方法非常宝贵。