Zhu Wenjing, Zhang Qiliang, Liu Min, Yan Meixing, Chu Xiao, Li Yongchun
Department of Pharmacy, School of Medicine, Qingdao Municipal Hospital, Qingdao University, Qingdao, 266011, Shandong, China.
Department of Orthopedics and Sports Medicine and Joint Surgery, Qingdao Municipal Hospital, Qingdao, Shandong, China.
Cancer Cell Int. 2021 Jan 30;21(1):81. doi: 10.1186/s12935-021-01779-1.
Liver cancer (LC) is one of the most fatal cancers throughout the world. More efficient and sensitive gene signatures that could accurately predict survival in LC patients are vitally needed to promote a better individualized and effective treatment.
MATERIAL/METHODS: 422 LC and adjacent normal tissues with both RNA-Seq and clinical data in TCGA were embedded in our study. Gene set enrichment analysis (GSEA) was applied to identify genes and hallmark gene sets that are more valuable for liver cancer therapy. Cox regression analysis was used to identify genes related to overall survival (OS) and build the prediction model. cBioPortal database was used to examine the alterations of the panel mRNA signature. ROC curves and Kaplan-Meier curves were used to validate the prediction model. Besides, the expression of the genes in the model were validated using quantitative real-time PCR in clinical tissue specimens.
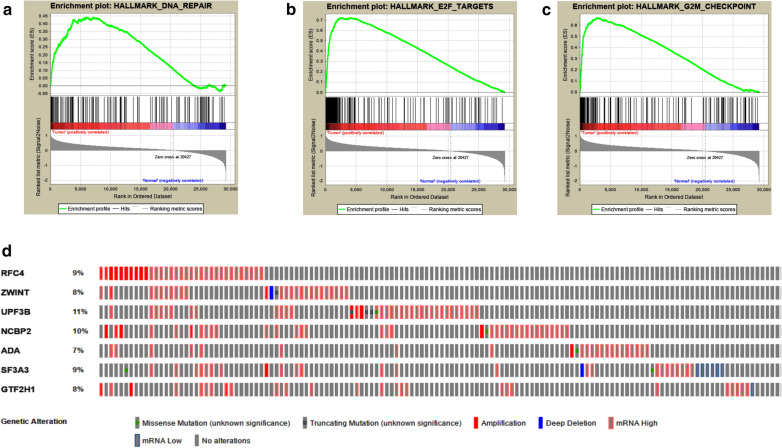
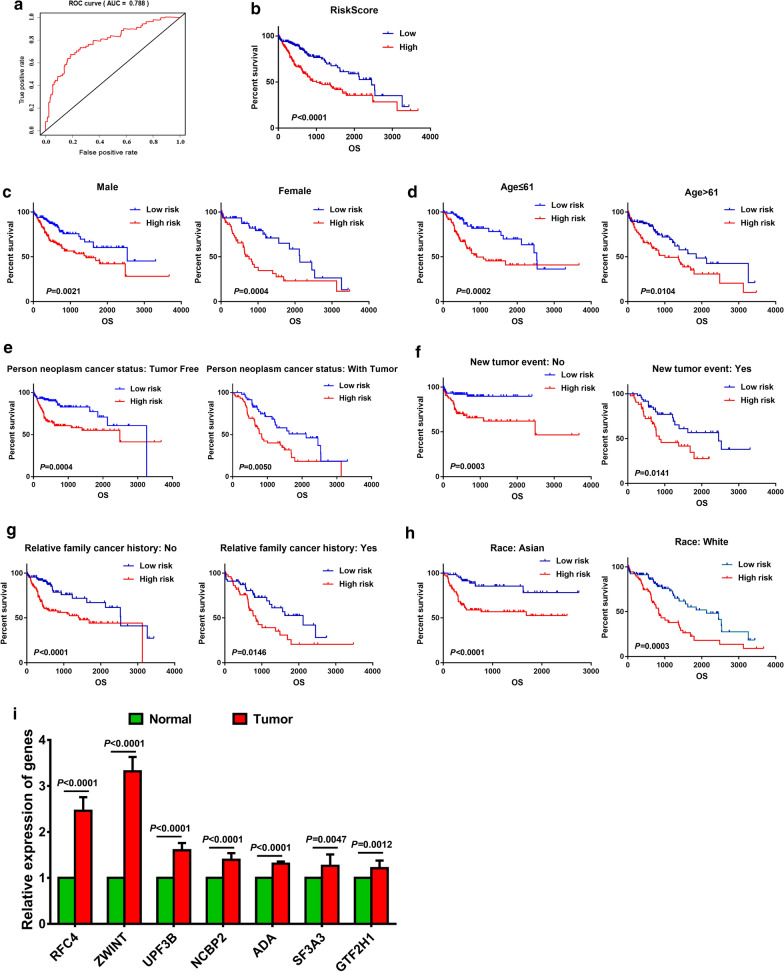
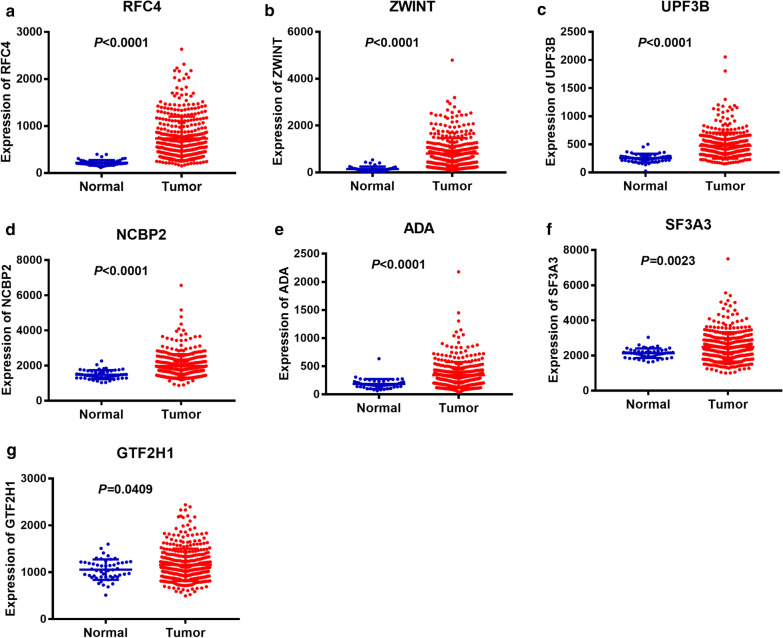
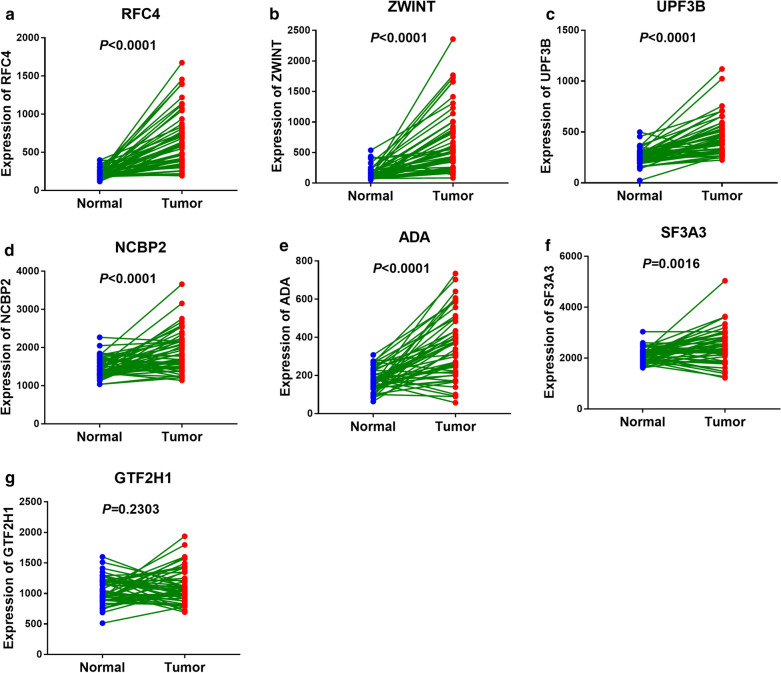
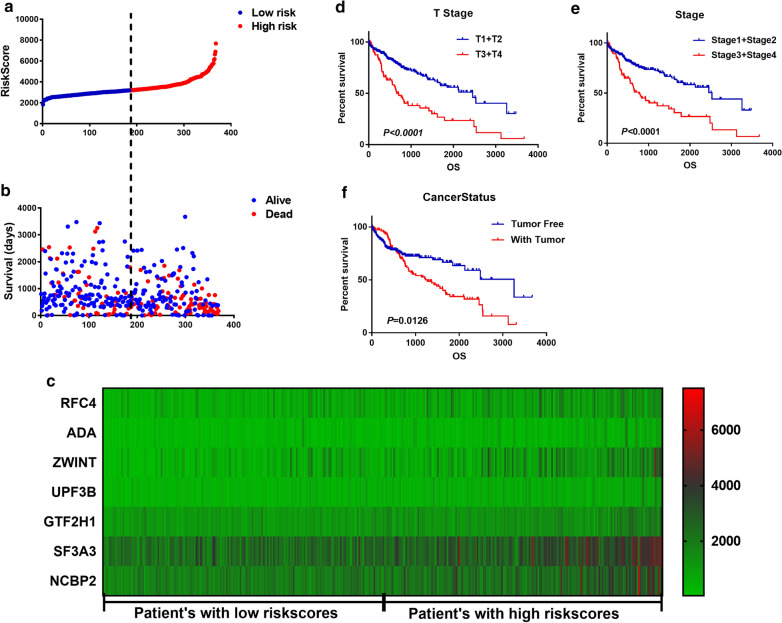
The panel of DNA repair-related mRNA signature consisted of seven mRNAs: RFC4 (replication factor C subunit 4), ZWINT (ZW10 interacting kinetochore protein), UPF3B (UPF3B regulator of nonsense mediated mRNA decay), NCBP2 (nuclear cap binding protein subunit 2), ADA (adenosine deaminase), SF3A3 (splicing factor 3a subunit 3) and GTF2H1 (general transcription factor IIH subunit 1). On-line analysis of cBioPortal database found that the expression of the panel mRNA has a wide variation ranging from 7 to 10%. All the mRNAs were significantly upregulated in LC tissues compared to normal tissues (P < 0.05). The risk model is closely related to the OS of LC patients. The hazard ratio (HR) is 2.184 [95% CI (confidence interval) 1.523-3.132] and log-rank P-value < 0.0001. For clinical specimen validation, we found that all of the genes in the model upregulated in liver cancer tissues versus normal liver tissues, which was consistent with the results predicted.
Our study demonstrated a mRNA signature including seven mRNA for prognosis prediction of LC. This panel gene signature provides a new criterion for accurate diagnosis and therapeutic target of LC.
肝癌(LC)是全球最致命的癌症之一。迫切需要更有效、更敏感的基因特征,以准确预测LC患者的生存情况,从而促进更好的个体化和有效治疗。
材料/方法:本研究纳入了TCGA中422例具有RNA测序和临床数据的LC及癌旁正常组织。采用基因集富集分析(GSEA)来鉴定对肝癌治疗更有价值的基因和标志性基因集。使用Cox回归分析来鉴定与总生存期(OS)相关的基因并构建预测模型。利用cBioPortal数据库检查该mRNA特征谱的改变情况。采用ROC曲线和Kaplan-Meier曲线验证预测模型。此外,在临床组织标本中通过定量实时PCR验证模型中基因的表达。
DNA修复相关的mRNA特征谱由7种mRNA组成:RFC4(复制因子C亚基4)、ZWINT(ZW10相互作用的动粒蛋白)、UPF3B(无义介导的mRNA衰变调节因子UPF3B)、NCBP2(核帽结合蛋白亚基2)、ADA(腺苷脱氨酶)、SF3A3(剪接因子3a亚基3)和GTF2H1(通用转录因子IIH亚基1)。cBioPortal数据库的在线分析发现,该mRNA特征谱的表达变化范围广泛,为7%至10%。与正常组织相比,所有mRNA在LC组织中均显著上调(P < 0.05)。风险模型与LC患者的OS密切相关。风险比(HR)为2.184 [95%置信区间(CI)1.523 - 3.132],对数秩P值< 0.0001。对于临床标本验证,我们发现模型中的所有基因在肝癌组织中相对于正常肝组织均上调,这与预测结果一致。
我们的研究证明了一种包含7种mRNA的特征谱可用于LC的预后预测。该基因特征谱为LC的准确诊断和治疗靶点提供了新的标准。