Zhuang Xuehan, Ye Rui, So Man-Ting, Lam Wai-Yee, Karim Anwarul, Yu Michelle, Ngo Ngoc Diem, Cherny Stacey S, Tam Paul Kwong-Hang, Garcia-Barcelo Maria-Mercè, Tang Clara Sze-Man, Sham Pak Chung
Department of Surgery, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China.
Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China.
NAR Genom Bioinform. 2020 Sep 22;2(3):lqaa071. doi: 10.1093/nargab/lqaa071. eCollection 2020 Sep.
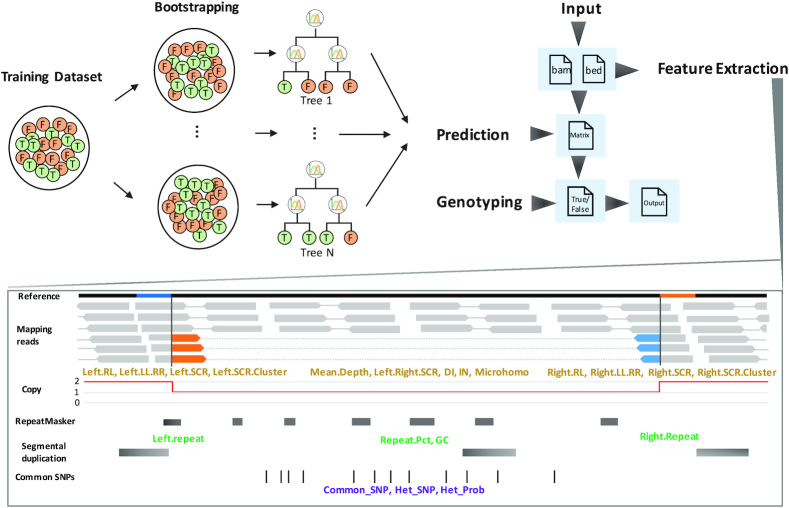
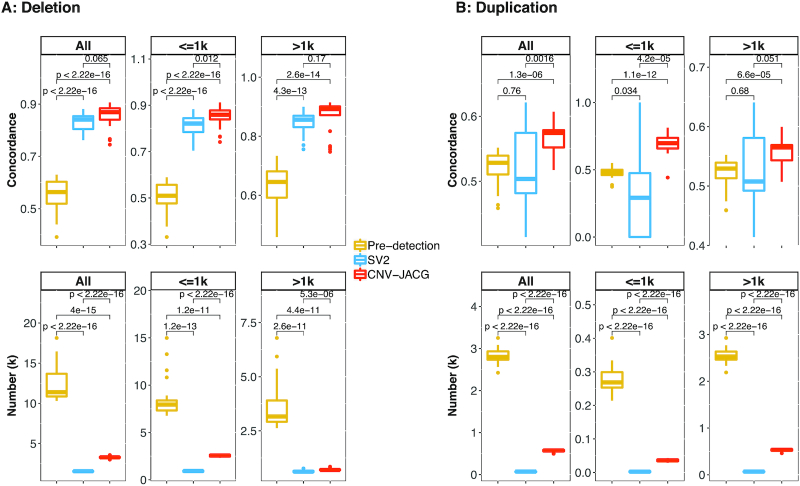
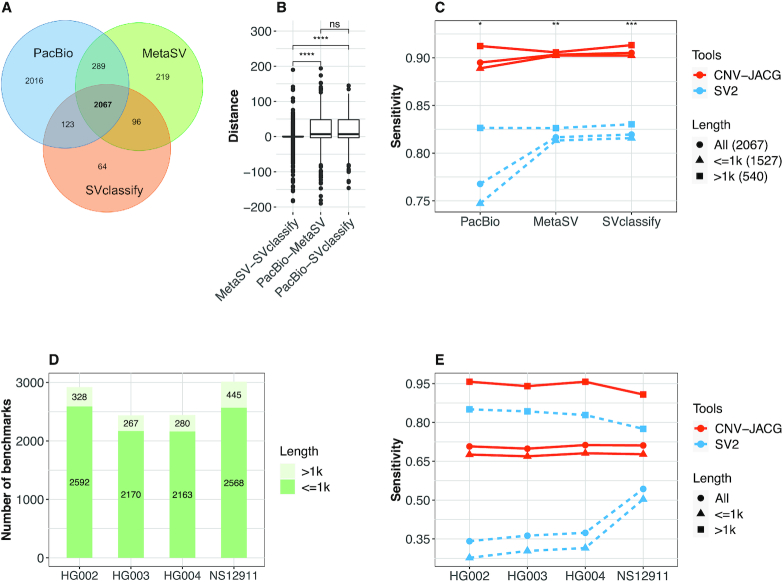
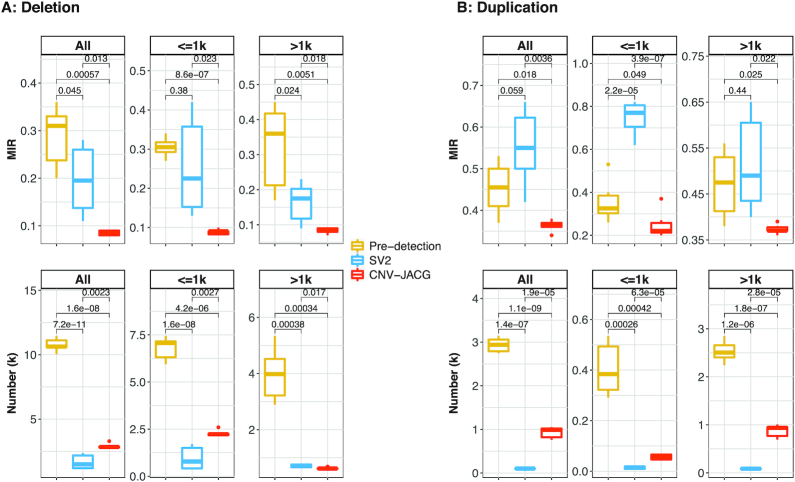
Detection of copy number variations (CNVs) is essential for uncovering genetic factors underlying human diseases. However, CNV detection by current methods is prone to error, and precisely identifying CNVs from paired-end whole genome sequencing (WGS) data is still challenging. Here, we present a framework, CNV-JACG, for udging the ccuracy of NVs and enotyping using paired-end WGS data. CNV-JACG is based on a random forest model trained on 21 distinctive features characterizing the CNV region and its breakpoints. Using the data from the 1000 Genomes Project, Genome in a Bottle Consortium, the Human Genome Structural Variation Consortium and in-house technical replicates, we show that CNV-JACG has superior sensitivity over the latest genotyping method, SV, particularly for the small CNVs (≤1 kb). We also demonstrate that CNV-JACG outperforms SV in terms of Mendelian inconsistency in trios and concordance between technical replicates. Our study suggests that CNV-JACG would be a useful tool in assessing the accuracy of CNVs to meet the ever-growing needs for uncovering the missing heritability linked to CNVs.
检测拷贝数变异(CNV)对于揭示人类疾病的遗传因素至关重要。然而,目前通过现有方法进行CNV检测容易出错,并且从双末端全基因组测序(WGS)数据中准确识别CNV仍然具有挑战性。在此,我们提出了一个名为CNV-JACG的框架,用于使用双末端WGS数据判断CNV的准确性并进行基因分型。CNV-JACG基于一个随机森林模型,该模型通过表征CNV区域及其断点的21个独特特征进行训练。利用来自千人基因组计划、基因组瓶子联盟、人类基因组结构变异联盟的数据以及内部技术重复数据,我们表明CNV-JACG比最新的基因分型方法SV具有更高的灵敏度,特别是对于小的CNV(≤1 kb)。我们还证明,在三人组中的孟德尔不一致性以及技术重复之间的一致性方面,CNV-JACG优于SV。我们的研究表明,CNV-JACG将成为评估CNV准确性的有用工具,以满足不断增长的揭示与CNV相关的缺失遗传力的需求。