Badri Michelle, Kurtz Zachary D, Bonneau Richard, Müller Christian L
Department of Biology, New York University, New York, NY 10012, USA.
Lodo Therapeutics, New York, NY 10016, USA.
NAR Genom Bioinform. 2020 Dec 17;2(4):lqaa100. doi: 10.1093/nargab/lqaa100. eCollection 2020 Dec.
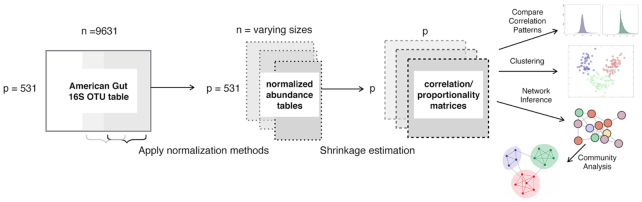
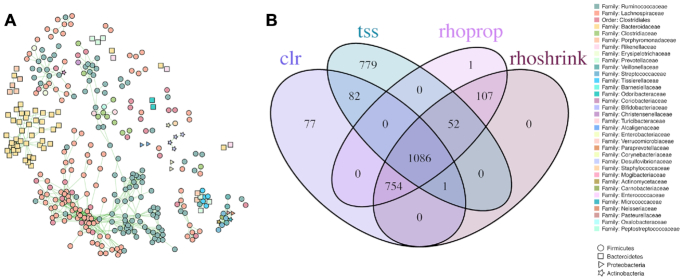
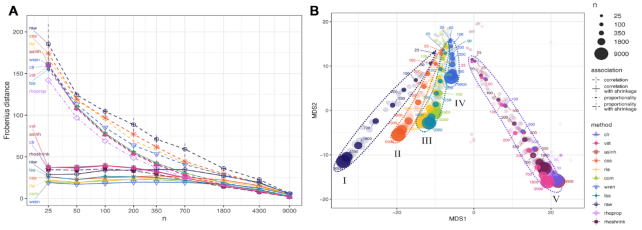
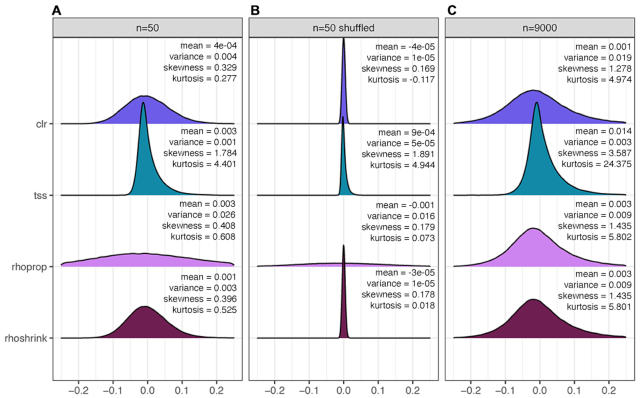
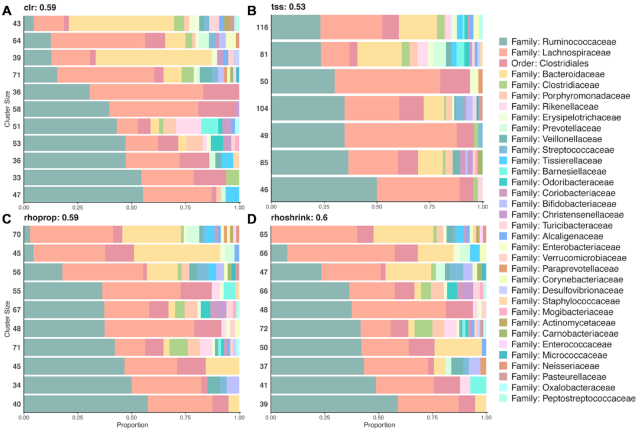
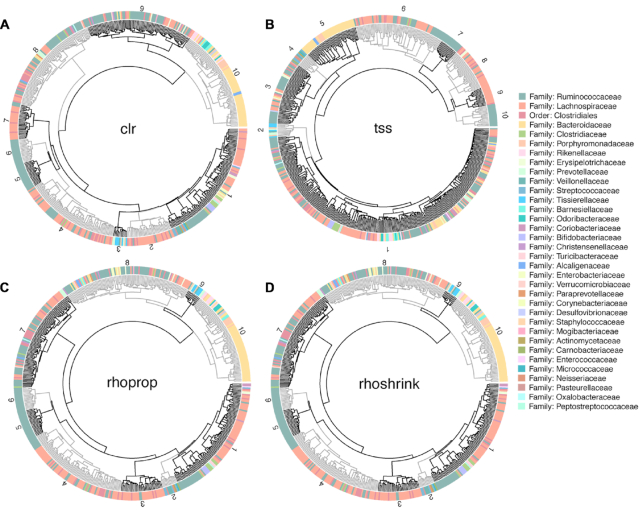
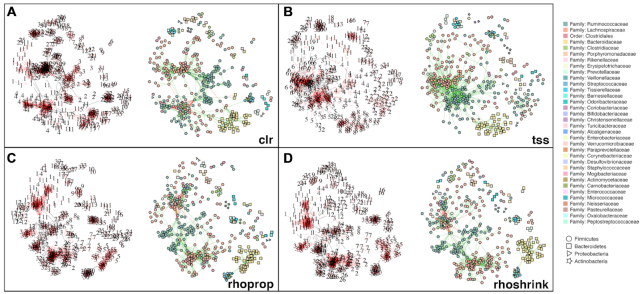
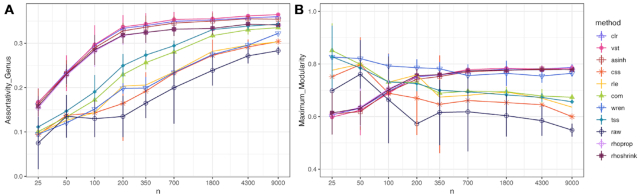
Estimation of statistical associations in microbial genomic survey count data is fundamental to microbiome research. Experimental limitations, including count compositionality, low sample sizes and technical variability, obstruct standard application of association measures and require data normalization prior to statistical estimation. Here, we investigate the interplay between data normalization, microbial association estimation and available sample size by leveraging the large-scale American Gut Project (AGP) survey data. We analyze the statistical properties of two prominent linear association estimators, correlation and proportionality, under different sample scenarios and data normalization schemes, including RNA-seq analysis workflows and log-ratio transformations. We show that shrinkage estimation, a standard statistical regularization technique, can universally improve the quality of taxon-taxon association estimates for microbiome data. We find that large-scale association patterns in the AGP data can be grouped into five normalization-dependent classes. Using microbial association network construction and clustering as downstream data analysis examples, we show that variance-stabilizing and log-ratio approaches enable the most taxonomically and structurally coherent estimates. Taken together, the findings from our reproducible analysis workflow have important implications for microbiome studies in multiple stages of analysis, particularly when only small sample sizes are available.
在微生物基因组调查计数数据中估计统计关联是微生物组研究的基础。实验限制,包括计数的组成性、小样本量和技术变异性,阻碍了关联度量的标准应用,并且在进行统计估计之前需要对数据进行归一化处理。在此,我们通过利用大规模的美国肠道项目(AGP)调查数据,研究数据归一化、微生物关联估计和可用样本量之间的相互作用。我们分析了两种著名的线性关联估计器(相关性和比例性)在不同样本场景和数据归一化方案下的统计特性,包括RNA测序分析工作流程和对数比变换。我们表明,收缩估计作为一种标准的统计正则化技术,可以普遍提高微生物组数据中分类单元-分类单元关联估计的质量。我们发现,AGP数据中的大规模关联模式可以分为五个依赖于归一化的类别。以微生物关联网络构建和聚类作为下游数据分析示例,我们表明方差稳定和对数比方法能够实现最具分类学和结构一致性的估计。总之,我们可重复分析工作流程的研究结果对微生物组研究多个分析阶段具有重要意义,特别是在只有小样本量可用的情况下。