Landles Christian, Milton Rebecca E, Jean Alexandre, McLarnon Stuart, McAteer Sean J, Taxy Bridget A, Osborne Georgina F, Zhang Chuangchuang, Duan Wenzhen, Howland David, Bates Gillian P
Department of Neurodegenerative Disease, Huntington's Disease Centre, UK Dementia Research Institute at UCL, Queen Square Institute of Neurology, University College London, London, UK.
Perkin Elmer Inc., Seer Green, UK.
Brain Commun. 2021 Jan 5;3(1):fcaa231. doi: 10.1093/braincomms/fcaa231. eCollection 2021.
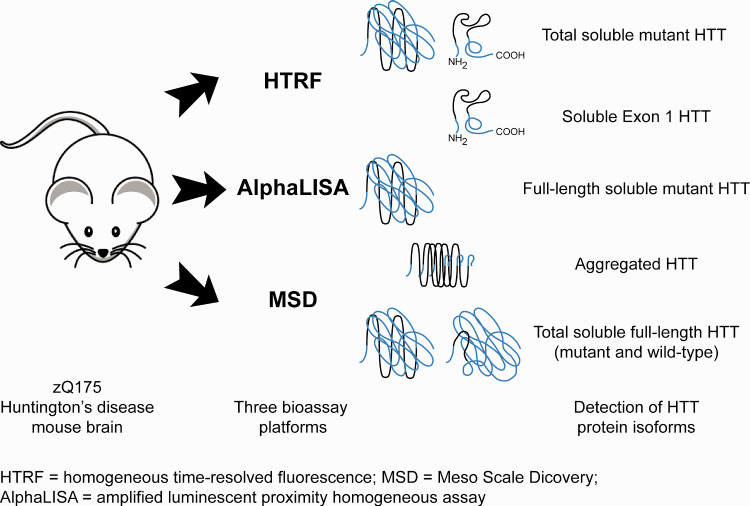
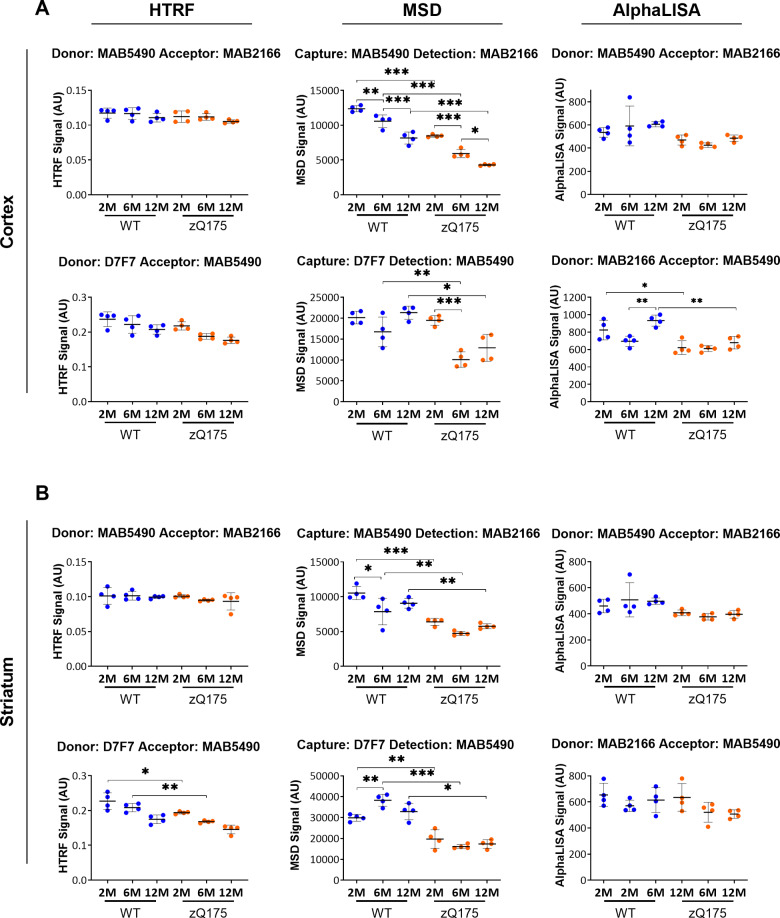
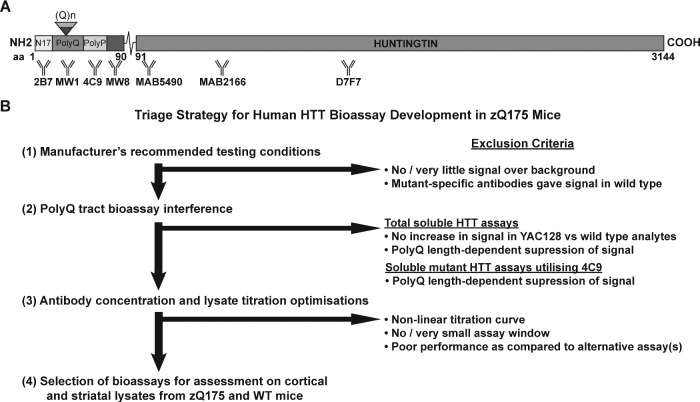
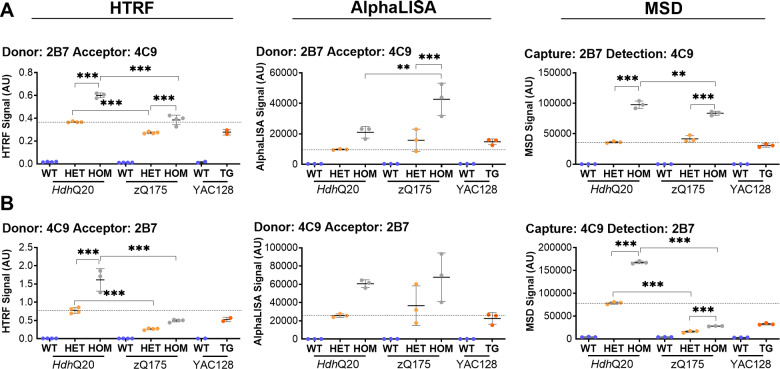
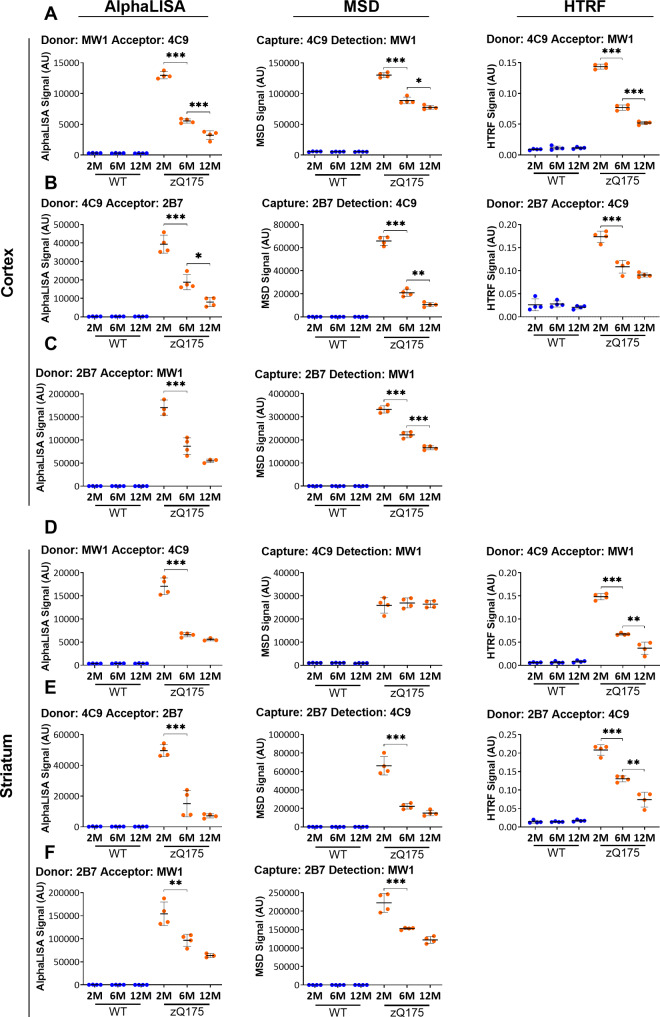
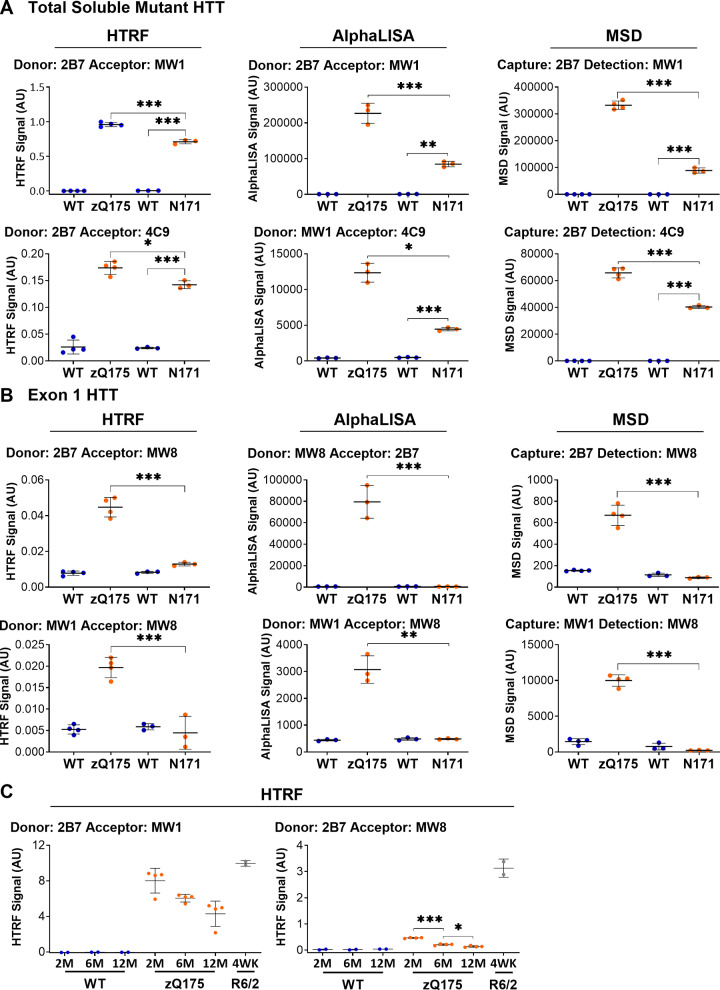
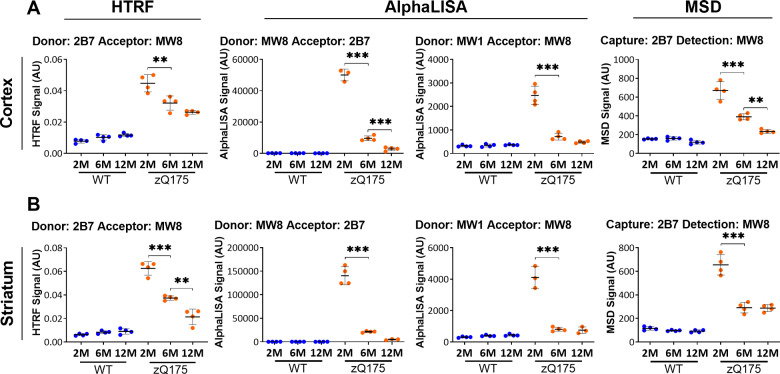
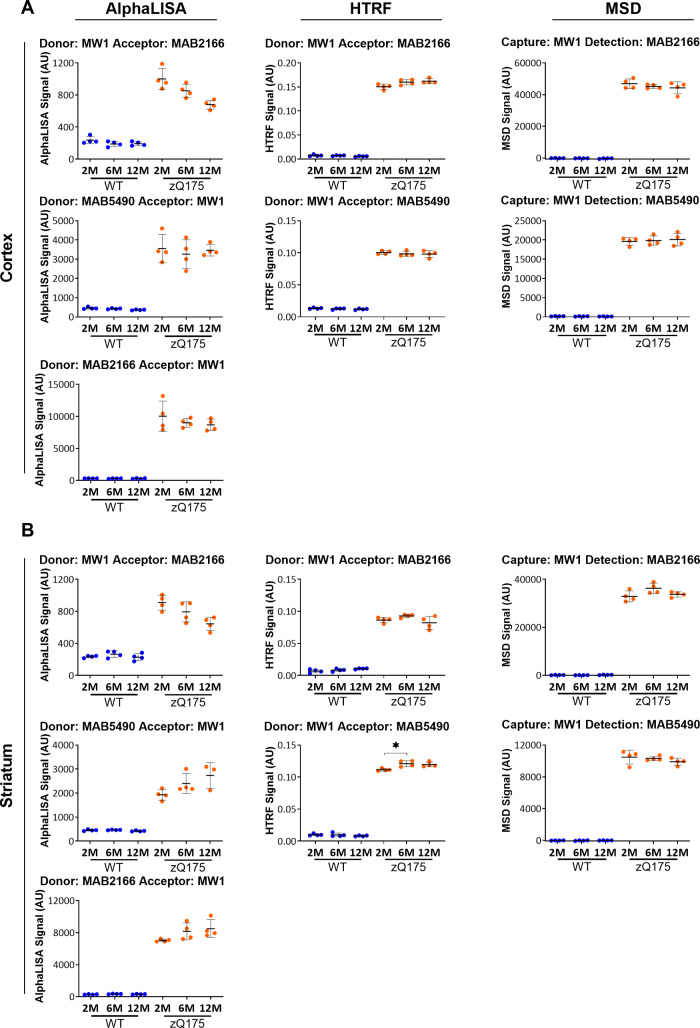
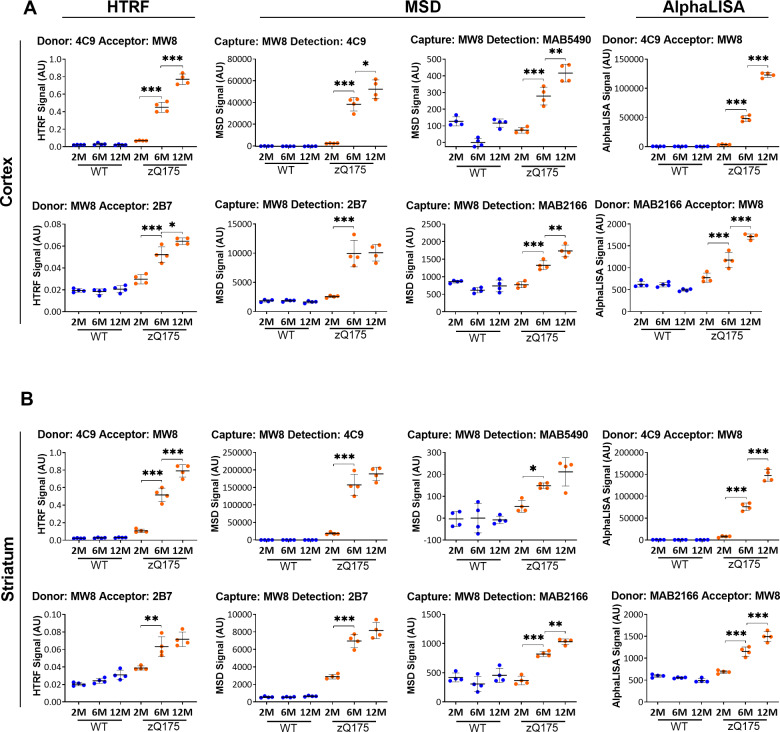
Huntington's disease is caused by a CAG / polyglutamine repeat expansion. Mutated CAG repeats undergo somatic instability, resulting in tracts of several hundred CAGs in the brain; and genetic modifiers of Huntington's disease have indicated that somatic instability is a major driver of age of onset and disease progression. As the CAG repeat expands, the likelihood that exon 1 does not splice to exon 2 increases, resulting in two transcripts that encode full-length huntingtin protein, as well as the highly pathogenic and aggregation-prone exon 1 huntingtin protein. Strategies that target the huntingtin gene or transcripts are a major focus of therapeutic development. It is essential that the levels of all isoforms of huntingtin protein can be tracked, to better understand the molecular pathogenesis, and to assess the impact of huntingtin protein-lowering approaches in preclinical studies and clinical trials. Huntingtin protein bioassays for soluble and aggregated forms of huntingtin protein are in widespread use on the homogeneous time-resolved fluorescence and Meso Scale Discovery platforms, but these do not distinguish between exon 1 huntingtin protein and full-length huntingtin protein. In addition, they are frequently used to quantify huntingtin protein levels in the context of highly expanded polyglutamine tracts, for which appropriate protein standards do not currently exist. Here, we set out to develop novel huntingtin protein bioassays to ensure that all soluble huntingtin protein isoforms could be distinguished. We utilized the zQ175 Huntington's disease mouse model that has ∼190 CAGs, a CAG repeat size for which protein standards are not available. Initially, 30 combinations of six antibodies were tested on three technology platforms: homogeneous time-resolved fluorescence, amplified luminescent proximity homogeneous assay and Meso Scale Discovery, and a triage strategy was employed to select the best assays. We found that, without a polyglutamine-length-matched standard, the vast majority of soluble mutant huntingtin protein assays cannot be used for quantitative purposes, as the highly expanded polyglutamine tract decreased assay performance. The combination of our novel assays, with those already in existence, provides a tool-kit to track: total soluble mutant huntingtin protein, soluble exon 1 huntingtin protein, soluble mutant huntingtin protein (excluding the exon 1 huntingtin protein) and total soluble full-length huntingtin protein (mutant and wild type). Several novel aggregation assays were also developed that track with disease progression. These selected assays can be used to compare the levels of huntingtin protein isoforms in a wide variety of mouse models of Huntington's disease and to determine how these change in response to genetic or therapeutic manipulations.
亨廷顿舞蹈症由CAG/多聚谷氨酰胺重复序列扩增引起。突变的CAG重复序列会发生体细胞不稳定性,导致大脑中出现数百个CAG序列片段;而亨廷顿舞蹈症的基因修饰因子表明,体细胞不稳定性是发病年龄和疾病进展的主要驱动因素。随着CAG重复序列的扩增,外显子1不与外显子2剪接的可能性增加,从而产生两种转录本,一种编码全长亨廷顿蛋白,另一种是具有高致病性且易于聚集的外显子1亨廷顿蛋白。针对亨廷顿基因或转录本的策略是治疗开发的主要重点。必须能够追踪亨廷顿蛋白所有异构体的水平,以便更好地理解分子发病机制,并评估在临床前研究和临床试验中降低亨廷顿蛋白方法的影响。用于检测可溶性和聚集形式亨廷顿蛋白的生物测定法在均相时间分辨荧光和Meso Scale Discovery平台上广泛使用,但这些方法无法区分外显子1亨廷顿蛋白和全长亨廷顿蛋白。此外,它们经常用于在高度扩增的多聚谷氨酰胺序列背景下定量亨廷顿蛋白水平,而目前不存在合适的蛋白质标准品。在此,我们着手开发新型亨廷顿蛋白生物测定法,以确保能够区分所有可溶性亨廷顿蛋白异构体。我们利用了具有约190个CAG的zQ175亨廷顿舞蹈症小鼠模型,这种CAG重复序列大小没有可用的蛋白质标准品。最初,在三个技术平台上测试了六种抗体的30种组合:均相时间分辨荧光、放大发光邻近均相分析和Meso Scale Discovery,并采用了一种筛选策略来选择最佳测定法。我们发现,在没有多聚谷氨酰胺长度匹配标准品的情况下,绝大多数可溶性突变亨廷顿蛋白测定法无法用于定量目的,因为高度扩增的多聚谷氨酰胺序列会降低测定性能。我们的新型测定法与现有测定法相结合,提供了一套工具来追踪:总可溶性突变亨廷顿蛋白、可溶性外显子1亨廷顿蛋白、可溶性突变亨廷顿蛋白(不包括外显子1亨廷顿蛋白)和总可溶性全长亨廷顿蛋白(突变型和野生型)。还开发了几种新型聚集测定法,可追踪疾病进展。这些选定的测定法可用于比较多种亨廷顿舞蹈症小鼠模型中亨廷顿蛋白异构体的水平,并确定它们如何因基因或治疗操作而发生变化。