University of Sheffield, Sheffield, UK.
Newcastle University, Newcastle upon Tyne, UK.
Langenbecks Arch Surg. 2022 Mar;407(2):517-527. doi: 10.1007/s00423-021-02129-5. Epub 2021 Mar 2.
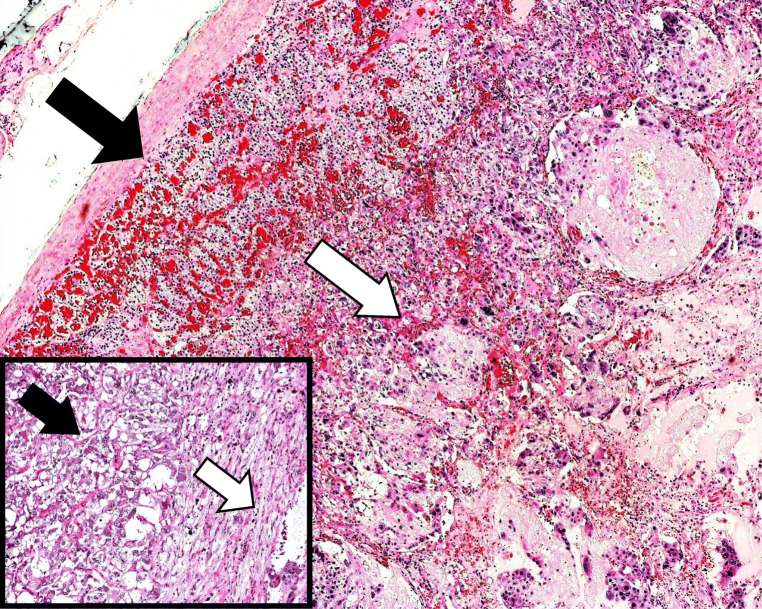
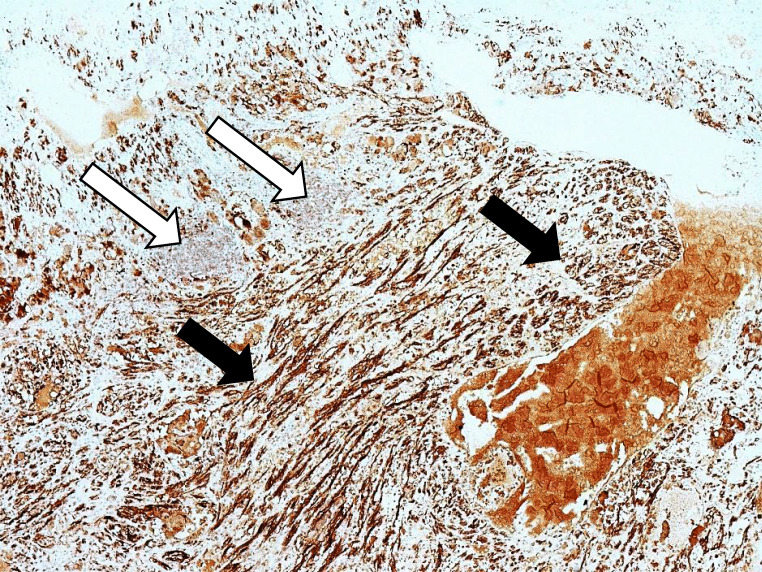
Composite phaeochromocytoma is a tumour containing a separate tumour of neuronal origin in addition to a chromaffin cell tumour. This study reports on two cases from a single centre's records and presents a systematic literature review of composite phaeochromocytomas.
In addition to describing 2 case reports, a systematic search of the Medline database from inception up to April 2020 was done for human case reports on composite phaeochromocytomas. Relevant titles and/or abstracts were screened, and full texts were reviewed to identify appropriate studies. Data was extracted and a descriptive analysis of presentation, clinical features, management strategies and outcomes was performed. The quality of included studies was assessed using a critical appraisal checklist.
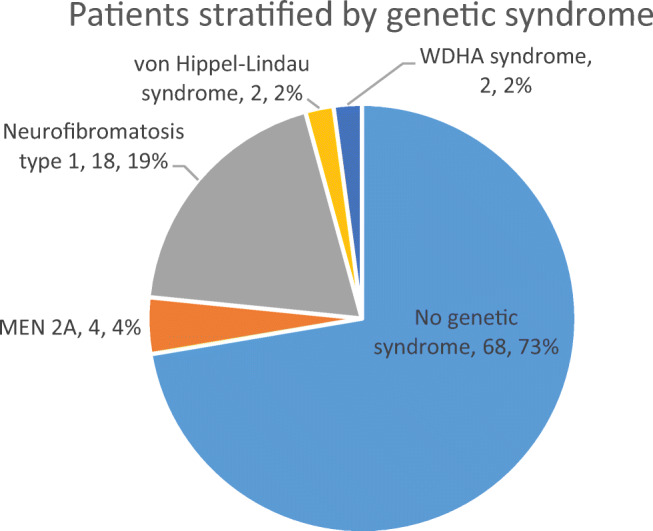
There were 62 studies included, with a total of 94 patients. Of 91 patients where data was available, the median (range) age of patients was 48 (4-86) years. Of 90 patients where information was provided, 57% were female. In at least 28% of patients, a genetic cause was identified. Common presenting features include abdominal pain, palpable mass, cardiovascular and gastrointestinal symptoms. The most common tumour component with phaeochromocytoma is ganglioneuroma; other components include ganglioneuroblastoma, neuroblastoma and malignant peripheral nerve sheath tumours. In patients with follow-up data (n=48), 85% of patients were alive and well at a median (range) follow-up time of 18 (0.5-168) months.
Composite phaeochromocytoma is a rare tumour, with a significant genetic predisposition. This review summarises available epidemiological data, which will be useful for clinicians managing this rare condition.
复合嗜铬细胞瘤是一种肿瘤,除嗜铬细胞瘤外,还含有单独起源于神经元的肿瘤。本研究报告了来自单一中心记录的两例病例,并对复合嗜铬细胞瘤进行了系统的文献回顾。
除描述 2 例病例报告外,还对从开始到 2020 年 4 月的 Medline 数据库进行了系统搜索,以查找关于复合嗜铬细胞瘤的人类病例报告。筛选了相关的标题和/或摘要,并对全文进行了回顾,以确定合适的研究。提取数据,并对表现、临床特征、管理策略和结果进行描述性分析。使用批判性评价检查表评估纳入研究的质量。
共纳入 62 项研究,共 94 例患者。在 91 例可获得数据的患者中,患者的中位(范围)年龄为 48(4-86)岁。在提供信息的 90 例患者中,57%为女性。在至少 28%的患者中,确定了遗传原因。常见的首发症状包括腹痛、可触及的肿块、心血管和胃肠道症状。与嗜铬细胞瘤最常见的肿瘤成分是神经节细胞瘤;其他成分包括神经节母细胞瘤、神经母细胞瘤和恶性周围神经鞘肿瘤。在有随访数据的患者(n=48)中,85%的患者在中位(范围)随访时间 18(0.5-168)个月时存活且状况良好。
复合嗜铬细胞瘤是一种罕见的肿瘤,具有显著的遗传易感性。本综述总结了现有的流行病学数据,这将对管理这种罕见疾病的临床医生有用。