Computational and Theoretical Chemistry Group, Department of Chemistry, Southern Methodist University, 3215 Daniel Avenue, Dallas, TX 75275-0314, USA.
Molecules. 2021 Feb 11;26(4):950. doi: 10.3390/molecules26040950.
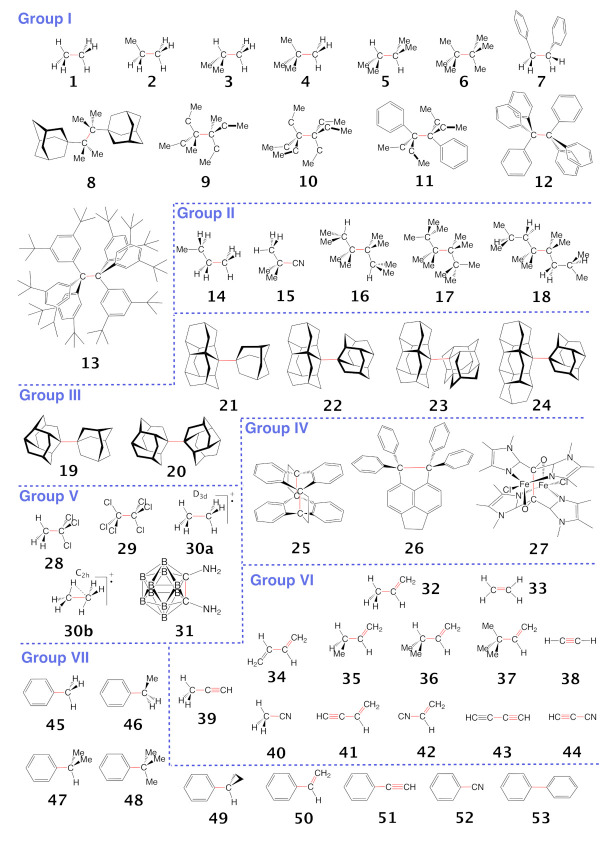
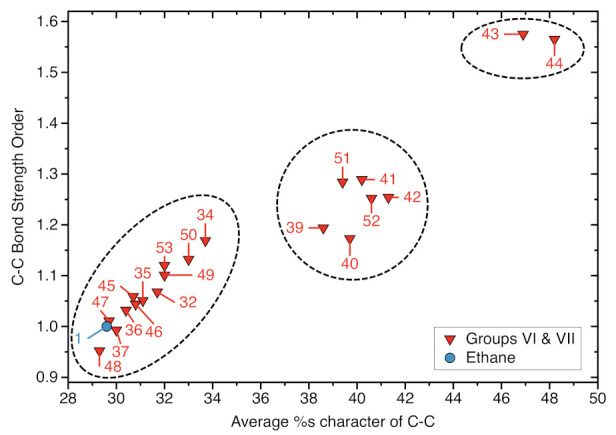
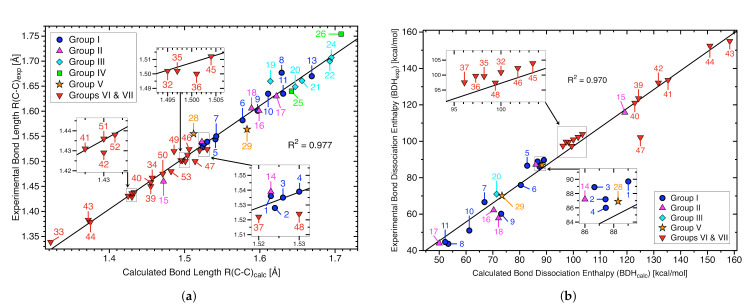
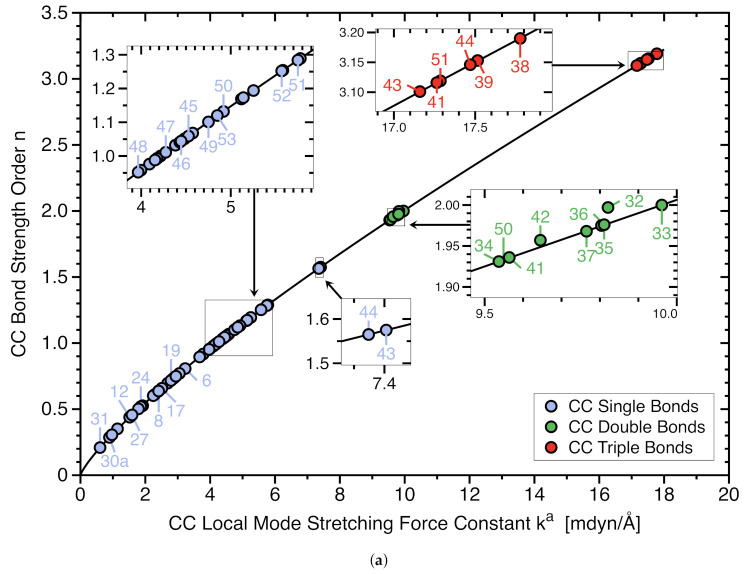
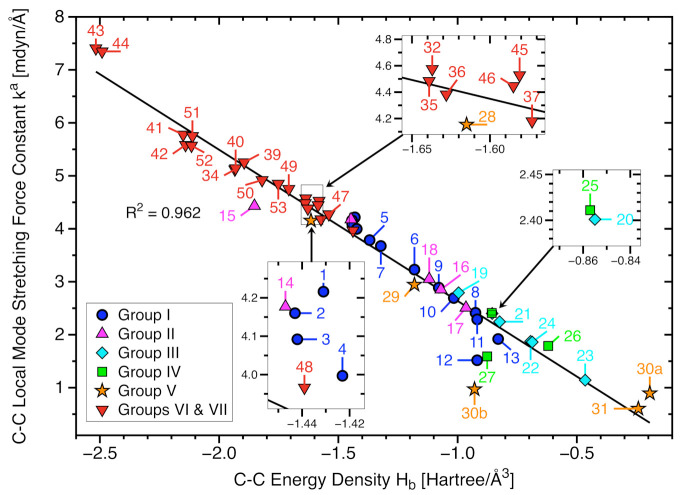
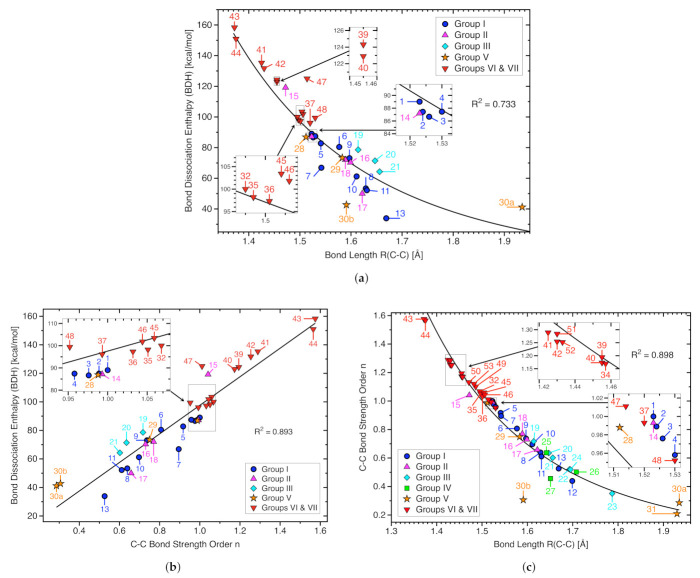
For decades one has strived to synthesize a compound with the longest covalent C-C bond applying predominantly steric hindrance and/or strain to achieve this goal. On the other hand electronic effects have been added to the repertoire, such as realized in the electron deficient ethane radical cation in its D3d form. Recently, negative hyperconjugation effects occurring in diamino-o-carborane analogs such as di-N,N-dimethylamino-o-carborane have been held responsible for their long C-C bonds. In this work we systematically analyzed CC bonding in a diverse set of 53 molecules including clamped bonds, highly sterically strained complexes such as diamondoid dimers, electron deficient species, and di-N,N-dimethylamino-o-carborane to cover the whole spectrum of possibilities for elongating a covalent C-C bond to the limit. As a quantitative intrinsic bond strength measure, we utilized local vibrational CC stretching force constants ka(CC) and related bond strength orders BSO (CC), computed at the ωB97X-D/aug-cc-pVTZ level of theory. Our systematic study quantifies for the first time that whereas steric hindrance and/or strain definitely elongate a C-C bond, electronic effects can lead to even longer and weaker C-C bonds. Within our set of molecules the electron deficient ethane radical cation, in D3d symmetry, acquires the longest C-C bond with a length of 1.935 Å followed by di-N,N-dimethylamino-o-carborane with a bond length of 1.930 Å. However, the C-C bond in di-N,N-dimethylamino-o-carborane is the weakest with a BSO value of 0.209 compared to 0.286 for the ethane radical cation; another example that the longer bond is not always the weaker bond. Based on our findings we provide new guidelines for the general characterization of CC bonds based on local vibrational CC stretching force constants and for future design of compounds with long C-C bonds.
几十年来,人们一直致力于合成具有最长共价 C-C 键的化合物,主要通过空间位阻和/或应变来实现这一目标。另一方面,电子效应也被加入到这个领域,例如在 D3d 形式的缺电子乙烷自由基阳离子中实现的电子效应。最近,在二氨基-o-卡硼烷类似物中出现的负超共轭效应,如二-N,N-二甲基氨基-o-卡硼烷,被认为是其长 C-C 键的原因。在这项工作中,我们系统地分析了包括夹键、高度空间应变复合物(如金刚石二聚体)、缺电子物种和二-N,N-二甲基氨基-o-卡硼烷在内的 53 种分子的 CC 键合,涵盖了拉长共价 C-C 键到极限的所有可能性。作为定量的内在键强度度量,我们利用局部振动 CC 伸缩力常数 ka(CC)和相关的键强度顺序 BSO(CC),在 ωB97X-D/aug-cc-pVTZ 理论水平上进行计算。我们的系统研究首次定量地表明,尽管空间位阻和/或应变肯定会延长 C-C 键,但电子效应也可能导致更长和更弱的 C-C 键。在我们的分子集合中,在 D3d 对称下,缺电子的乙烷自由基阳离子获得了最长的 C-C 键,长度为 1.935 Å,其次是二-N,N-二甲基氨基-o-卡硼烷,长度为 1.930 Å。然而,二-N,N-二甲基氨基-o-卡硼烷的 C-C 键是最弱的,BSO 值为 0.209,而乙烷自由基阳离子的 BSO 值为 0.286;这表明较长的键并不总是较弱的键。基于我们的发现,我们为基于局部振动 CC 伸缩力常数的 CC 键的一般特征化以及具有长 C-C 键的化合物的未来设计提供了新的指导方针。