Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, USA.
Genome Res. 2021 Apr;31(4):721-731. doi: 10.1101/gr.269613.120. Epub 2021 Mar 19.
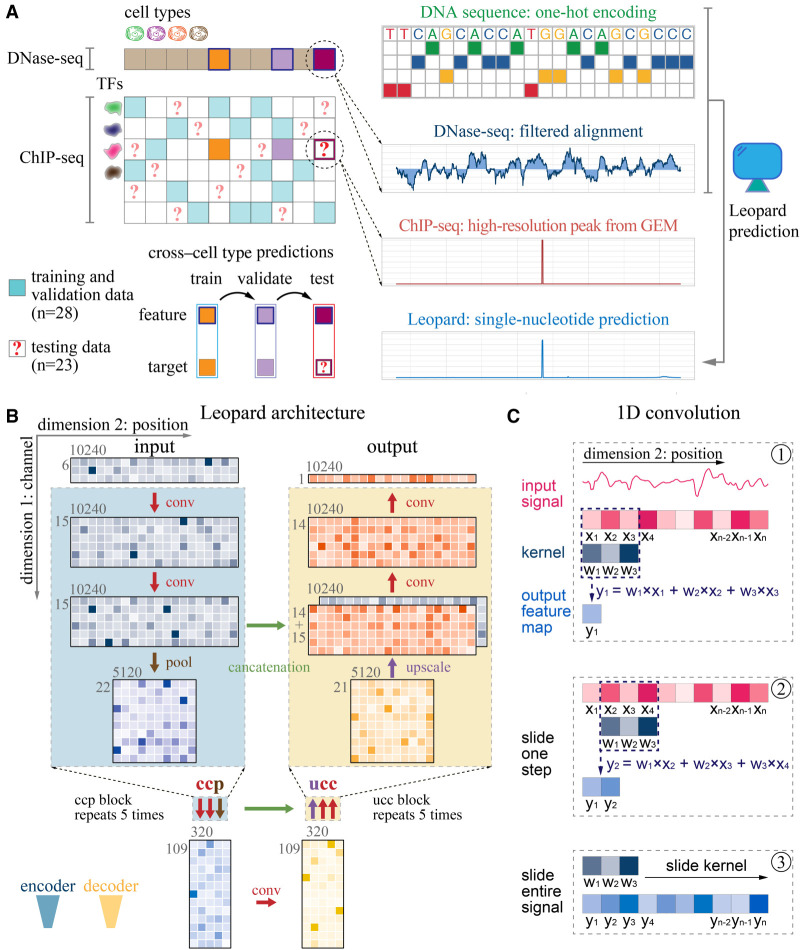
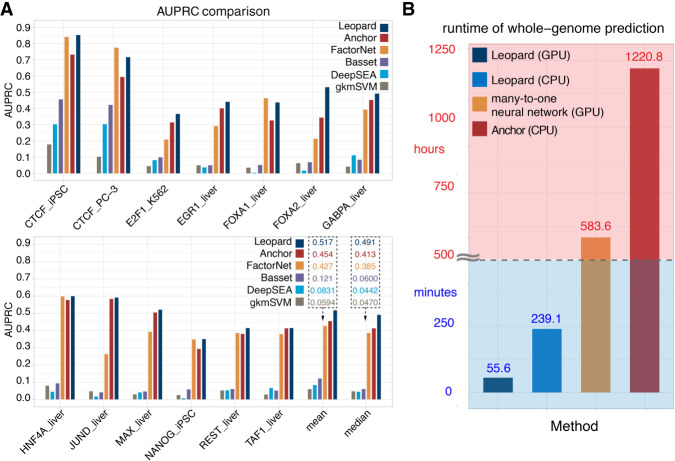
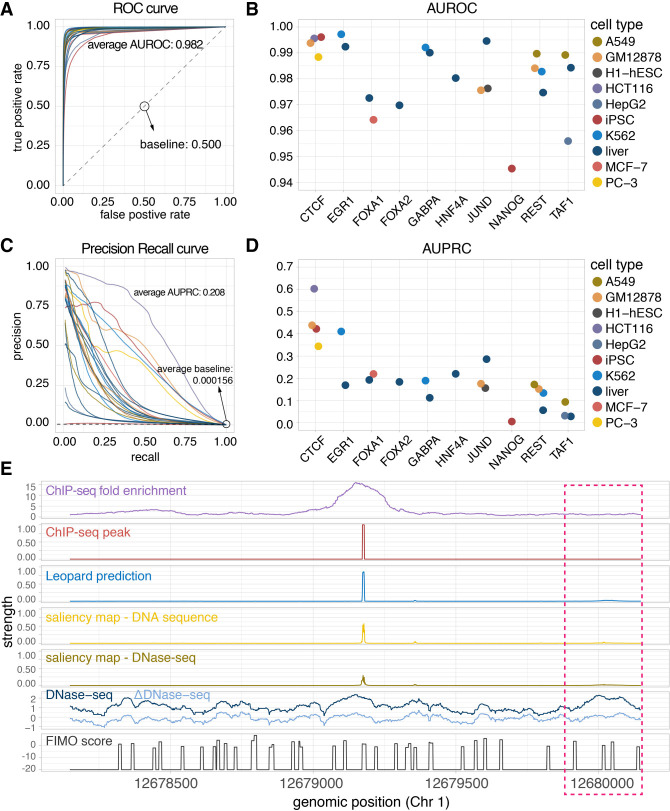
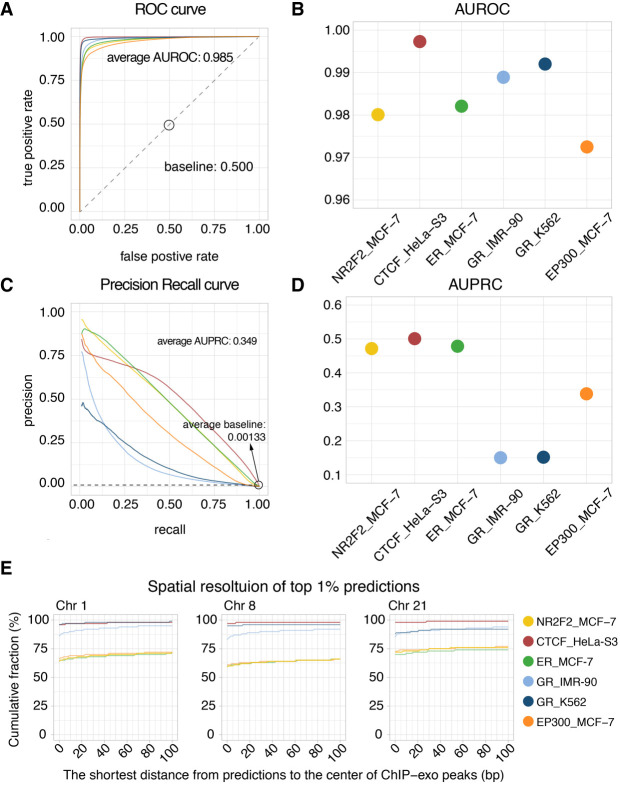
Decoding the cell type-specific transcription factor (TF) binding landscape at single-nucleotide resolution is crucial for understanding the regulatory mechanisms underlying many fundamental biological processes and human diseases. However, limits on time and resources restrict the high-resolution experimental measurements of TF binding profiles of all possible TF-cell type combinations. Previous computational approaches either cannot distinguish the cell context-dependent TF binding profiles across diverse cell types or can only provide a relatively low-resolution prediction. Here we present a novel deep learning approach, Leopard, for predicting TF binding sites at single-nucleotide resolution, achieving the average area under receiver operating characteristic curve (AUROC) of 0.982 and the average area under precision recall curve (AUPRC) of 0.208. Our method substantially outperformed the state-of-the-art methods Anchor and FactorNet, improving the predictive AUPRC by 19% and 27%, respectively, when evaluated at 200-bp resolution. Meanwhile, by leveraging a many-to-many neural network architecture, Leopard features a hundredfold to thousandfold speedup compared with current many-to-one machine learning methods.
解析单细胞转录因子(TF)结合图谱的核苷酸分辨率对于理解许多基本生物学过程和人类疾病的调控机制至关重要。然而,时间和资源的限制限制了所有可能的 TF-细胞类型组合的 TF 结合谱的高分辨率实验测量。以前的计算方法要么不能区分不同细胞类型中依赖于细胞环境的 TF 结合谱,要么只能提供相对较低分辨率的预测。在这里,我们提出了一种新的深度学习方法 Leopard,用于预测单核苷酸分辨率的 TF 结合位点,平均接收者操作特征曲线下面积(AUROC)为 0.982,平均精度召回曲线下面积(AUPRC)为 0.208。我们的方法大大优于最先进的方法 Anchor 和 FactorNet,当评估分辨率为 200 个碱基时,预测 AUPRC 分别提高了 19%和 27%。同时,通过利用多对多神经网络架构,与当前的多对一机器学习方法相比,Leopard 的速度提高了百倍到千倍。