Intensive Care Unit, The First Affiliated Hospital of Guangxi Medical University, Nanning, China.
School of Medicine, Southern University of Science and Technology, Shenzhen, China.
Front Cell Infect Microbiol. 2021 Mar 16;11:637018. doi: 10.3389/fcimb.2021.637018. eCollection 2021.
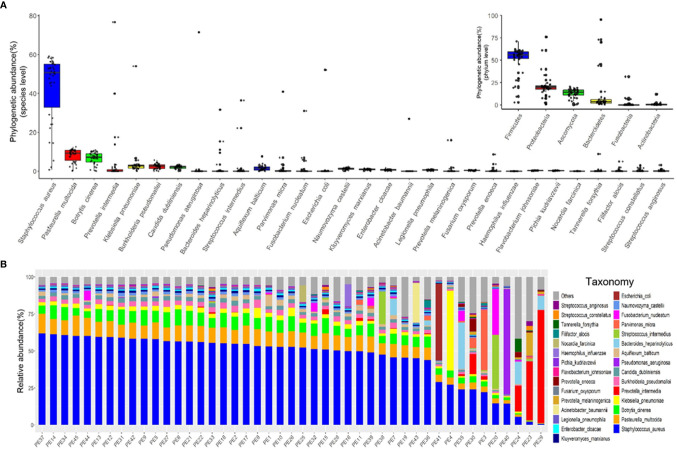
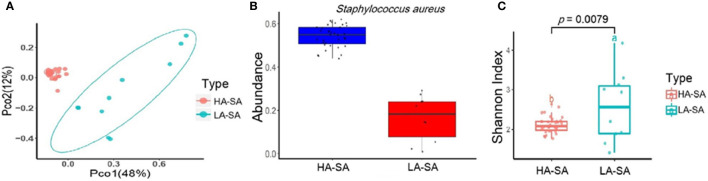
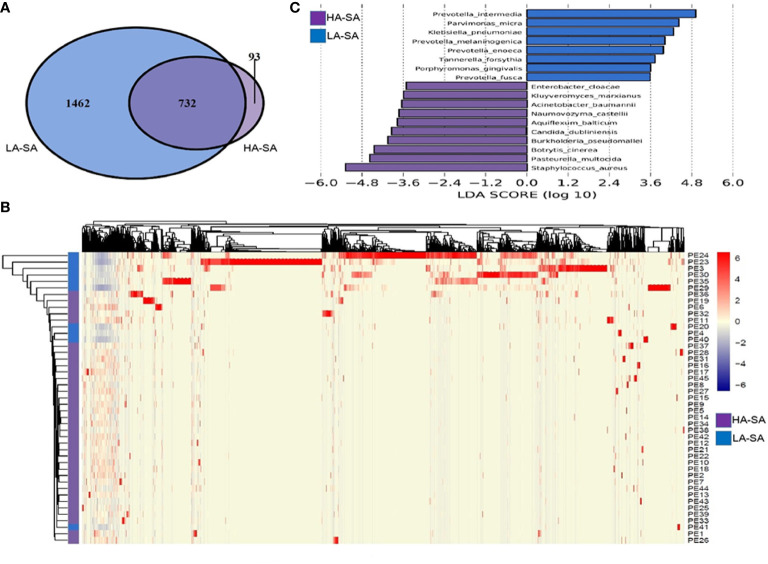
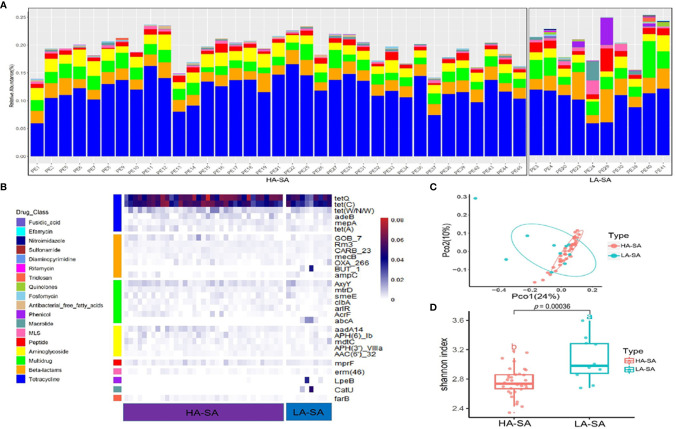
Identification of the offending organism and appropriate antimicrobial therapy are crucial for treating empyema. Diagnosis of empyema is largely obscured by the conventional bacterial cultivation and PCR process that has relatively low sensitivity, leading to limited understanding of the etiopathogenesis, microbiology, and role of antibiotics in the pleural cavity. To expand our understanding of its pathophysiology, we have carried out a metagenomic snapshot of the pleural effusion from 45 empyema patients by Illumina sequencing platform to assess its taxonomic, and antibiotic resistome structure. Our results showed that the variation of microbiota in the pleural effusion is generally stratified, not continuous. There are two distinct microbiome clusters observed in the forty-five samples: HA-SA type and LA-SA type. The categorization is mostly driven by species composition: HA-SA type is marked by as the core species, with other enriched 6 bacteria and 3 fungi, forming a low diversity and highly stable microbial community; whereas the LA-SA type has a more diverse microbial community with a distinct set of bacterial species that are assumed to be the oral origin. The microbial community does not shape the dominant antibiotic resistance classes which were common in the two types, while the increase of microbial diversity was correlated with the increase in antibiotic resistance genes. The existence of well-balanced microbial symbiotic states might respond differently to pathogen colonization and drug intake. This study provides a deeper understanding of the pathobiology of pleural empyema and suggests that potential resistance genes may hinder the antimicrobial therapy of empyema.
鉴定致病生物体和使用适当的抗菌药物治疗是治疗脓胸的关键。脓胸的诊断受到传统细菌培养和 PCR 过程的严重限制,这些方法的灵敏度相对较低,导致我们对其病因发病机制、微生物学以及抗生素在胸腔中的作用的理解有限。为了更深入地了解其病理生理学,我们通过 Illumina 测序平台对 45 例脓胸患者的胸腔积液进行了宏基因组快照分析,以评估其分类和抗生素耐药组结构。我们的研究结果表明,胸腔积液中的微生物群变化通常是分层的,而不是连续的。在这 45 个样本中观察到两种截然不同的微生物群落:HA-SA 型和 LA-SA 型。分类主要由物种组成驱动:HA-SA 型以 为核心物种,其他丰富的 6 种细菌和 3 种真菌,形成低多样性和高度稳定的微生物群落;而 LA-SA 型具有更丰富的微生物群落,其中包含一些假定来源于口腔的独特细菌物种。微生物群落并不决定两种类型中常见的主要抗生素耐药类群,而微生物多样性的增加与抗生素耐药基因的增加相关。平衡良好的微生物共生状态的存在可能会对病原体定植和药物摄入产生不同的反应。这项研究深入了解了脓胸的病理生物学,并提示潜在的耐药基因可能会阻碍脓胸的抗菌治疗。