Zhang Minzhe, Li Qiwei, Xie Yang
Quantitative Biomedical Research Center, Department of Clinical Sciences, U.T. Southwestern Medical Center, Dallas, TX 75390, USA.
Department of Bioinformatics, U.T. Southwestern Medical Center, Dallas, TX 75390, USA.
Quant Biol. 2018 Sep;6(3):275-286. doi: 10.1007/s40484-018-0149-2. Epub 2018 Aug 30.
The recently emerged technology of methylated RNA immunoprecipitation sequencing (MeRIP-seq) sheds light on the study of RNA epigenetics. This new bioinformatics question calls for effective and robust peaking calling algorithms to detect mRNA methylation sites from MeRIP-seq data.
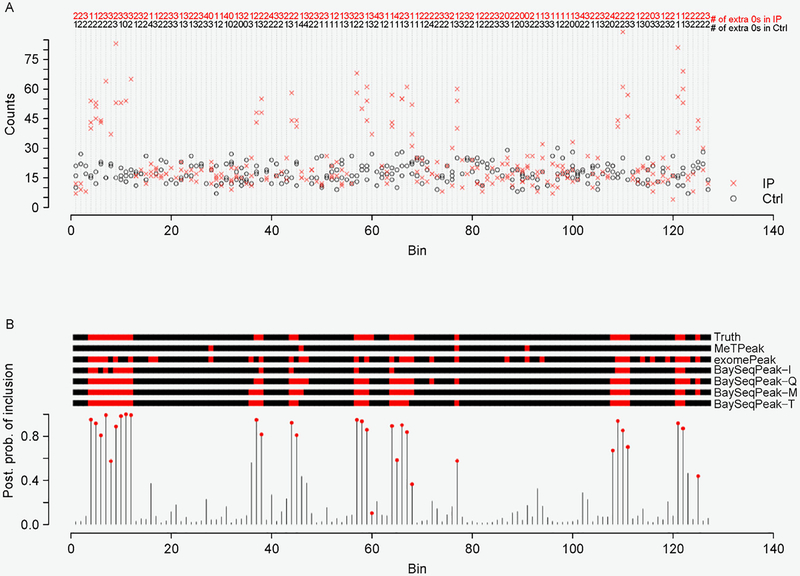
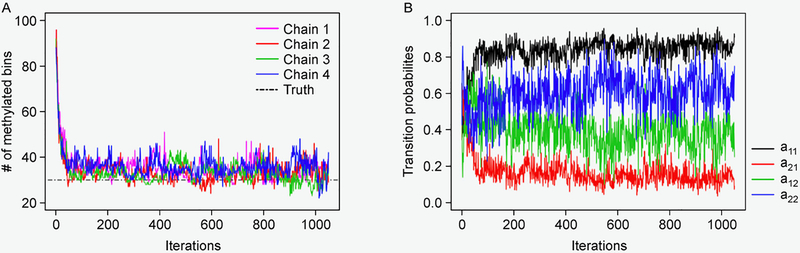
We propose a Bayesian hierarchical model to detect methylation sites from MeRIP-seq data. Our modeling approach includes several important characteristics. First, it models the zero-inflated and over-dispersed counts by deploying a zero-inflated negative binomial model. Second, it incorporates a hidden Markov model (HMM) to account for the spatial dependency of neighboring read enrichment. Third, our Bayesian inference allows the proposed model to borrow strength in parameter estimation, which greatly improves the model stability when dealing with MeRIP-seq data with a small number of replicates. We use Markov chain Monte Carlo (MCMC) algorithms to simultaneously infer the model parameters in a fashion. The R Shiny demo is available at https://qiwei.shinyapps.io/BaySeqPeak and the R/C ++ code is available at https://github.com/liqiwei2000/BaySeqPeak.
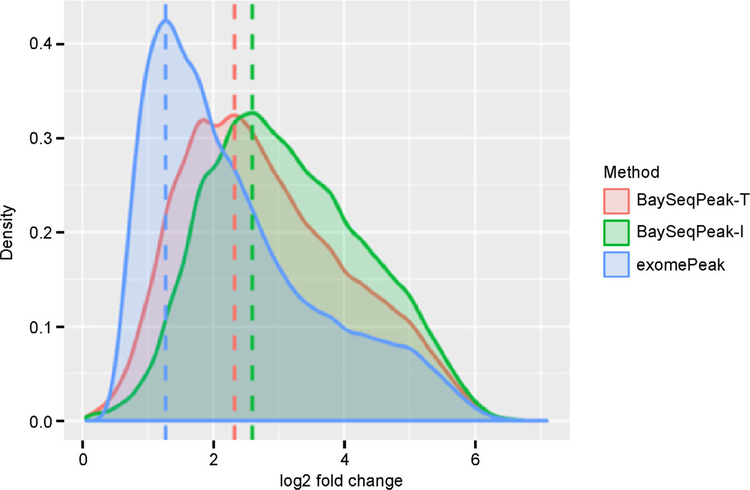
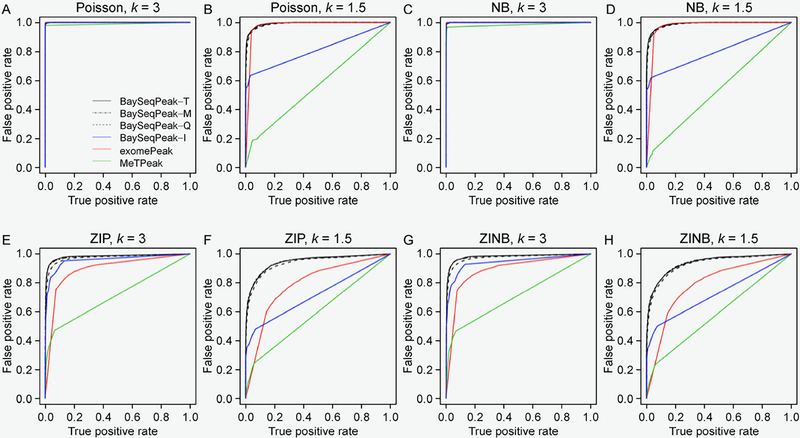
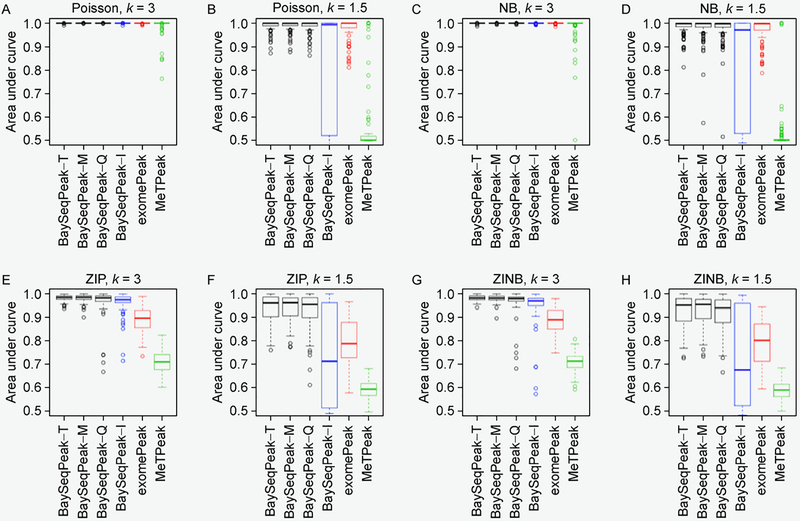
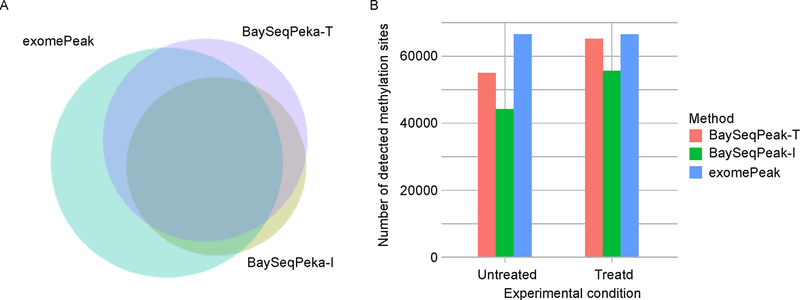
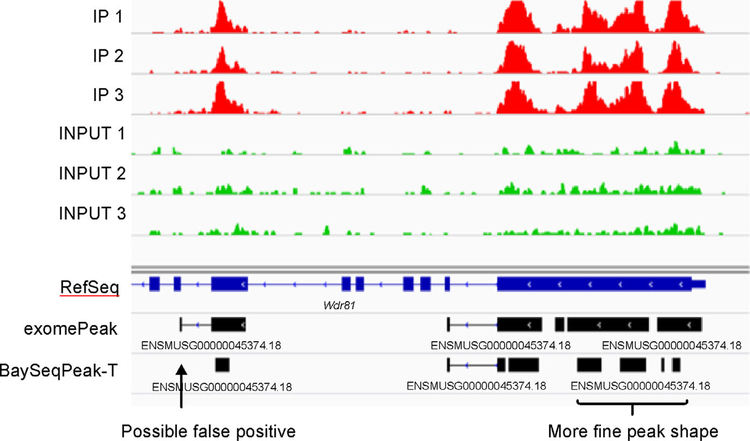
In simulation studies, the proposed method outperformed the competing methods exomePeak and MeTPeak, especially when an excess of zeros were present in the data. In real MeRIP-seq data analysis, the proposed method identified methylation sites that were more consistent with biological knowledge, and had better spatial resolution compared to the other methods.
In this study, we develop a Bayesian hierarchical model to identify methylation peaks in MeRIP-seq data. The proposed method has a competitive edge over existing methods in terms of accuracy, robustness and spatial resolution.
最近出现的甲基化RNA免疫沉淀测序(MeRIP-seq)技术为RNA表观遗传学研究提供了新的视角。这个新的生物信息学问题需要有效且强大的峰检测算法,以便从MeRIP-seq数据中检测mRNA甲基化位点。
我们提出了一种贝叶斯层次模型来从MeRIP-seq数据中检测甲基化位点。我们的建模方法包括几个重要特征。首先,通过部署零膨胀负二项式模型对零膨胀和过度分散的计数进行建模。其次,纳入隐藏马尔可夫模型(HMM)以考虑相邻读数富集的空间依赖性。第三,我们的贝叶斯推理允许所提出的模型在参数估计中借用优势,这在处理少量重复的MeRIP-seq数据时大大提高了模型稳定性。我们使用马尔可夫链蒙特卡罗(MCMC)算法以一种方式同时推断模型参数。R Shiny演示可在https://qiwei.shinyapps.io/BaySeqPeak获得,R/C++代码可在https://github.com/liqiwei2000/BaySeqPeak获得。
在模拟研究中,所提出的方法优于竞争方法外显子峰(exomePeak)和甲基化峰检测方法(MeTPeak),特别是当数据中存在大量零时。在实际的MeRIP-seq数据分析中,所提出的方法识别出的甲基化位点与生物学知识更一致,并且与其他方法相比具有更好的空间分辨率。
在本研究中,我们开发了一种贝叶斯层次模型来识别MeRIP-seq数据中的甲基化峰。所提出的方法在准确性、稳健性和空间分辨率方面比现有方法具有竞争优势。