Department of Biostatistics, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, USA.
Department of Statistics and Data Science, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA.
Genome Res. 2021 Oct;31(10):1807-1818. doi: 10.1101/gr.268722.120. Epub 2021 Apr 9.
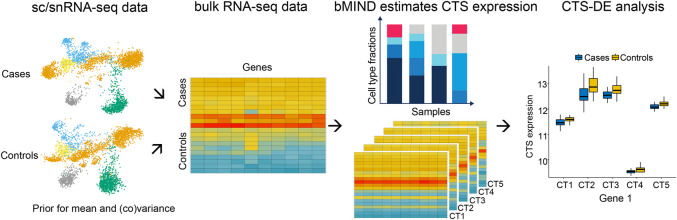
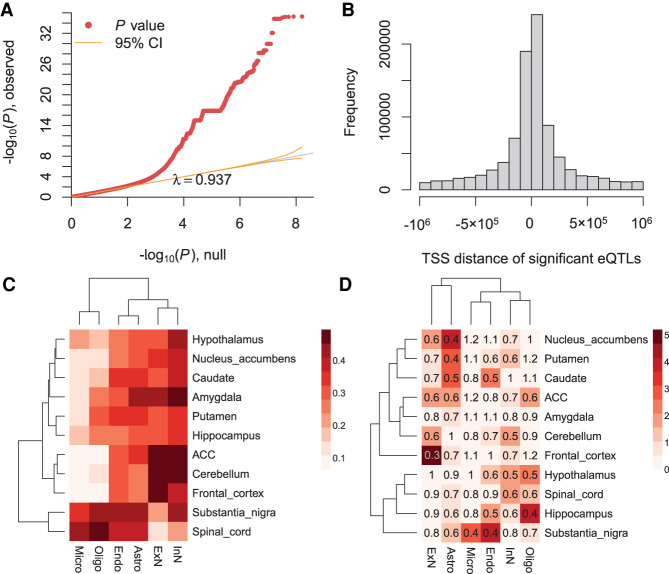
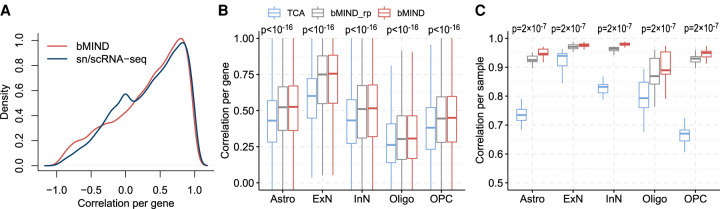
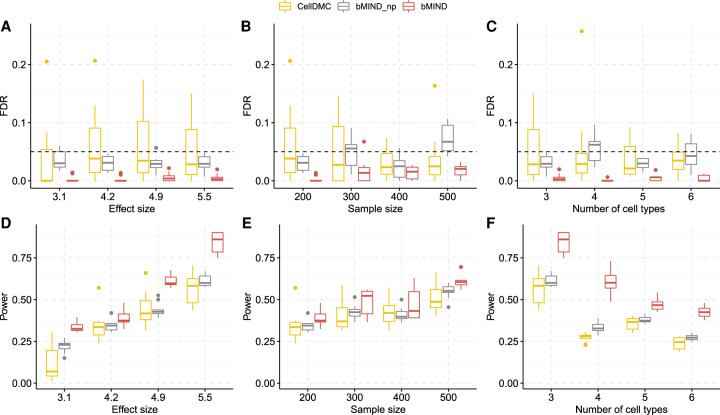
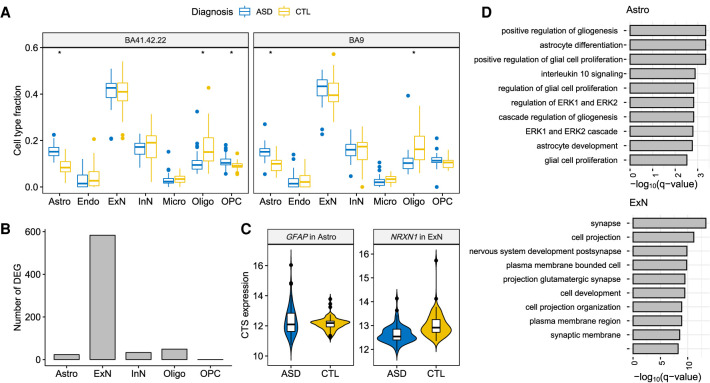
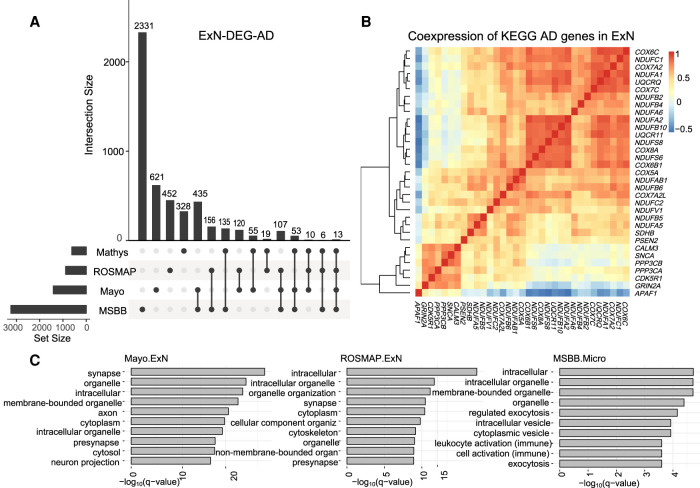
When assessed over a large number of samples, bulk RNA sequencing provides reliable data for gene expression at the tissue level. Single-cell RNA sequencing (scRNA-seq) deepens those analyses by evaluating gene expression at the cellular level. Both data types lend insights into disease etiology. With current technologies, scRNA-seq data are known to be noisy. Constrained by costs, scRNA-seq data are typically generated from a relatively small number of subjects, which limits their utility for some analyses, such as identification of gene expression quantitative trait loci (eQTLs). To address these issues while maintaining the unique advantages of each data type, we develop a Bayesian method (bMIND) to integrate bulk and scRNA-seq data. With a prior derived from scRNA-seq data, we propose to estimate sample-level cell type-specific (CTS) expression from bulk expression data. The CTS expression enables large-scale sample-level downstream analyses, such as detection of CTS differentially expressed genes (DEGs) and eQTLs. Through simulations, we show that bMIND improves the accuracy of sample-level CTS expression estimates and increases the power to discover CTS DEGs when compared to existing methods. To further our understanding of two complex phenotypes, autism spectrum disorder and Alzheimer's disease, we apply bMIND to gene expression data of relevant brain tissue to identify CTS DEGs. Our results complement findings for CTS DEGs obtained from snRNA-seq studies, replicating certain DEGs in specific cell types while nominating other novel genes for those cell types. Finally, we calculate CTS eQTLs for 11 brain regions by analyzing Genotype-Tissue Expression Project data, creating a new resource for biological insights.
当评估大量样本时,批量 RNA 测序可提供组织水平基因表达的可靠数据。单细胞 RNA 测序 (scRNA-seq) 通过评估细胞水平的基因表达来深化这些分析。这两种数据类型都有助于深入了解疾病的病因。目前的技术已知 scRNA-seq 数据存在噪声。受成本限制,scRNA-seq 数据通常是从相对较少的个体中生成的,这限制了它们在某些分析中的应用,例如鉴定基因表达数量性状基因座 (eQTL)。为了解决这些问题,同时保持每种数据类型的独特优势,我们开发了一种贝叶斯方法 (bMIND) 来整合批量和 scRNA-seq 数据。利用来自 scRNA-seq 数据的先验知识,我们建议从批量表达数据中估计样本水平的细胞类型特异性 (CTS) 表达。CTS 表达使大规模样本水平的下游分析成为可能,例如检测 CTS 差异表达基因 (DEG) 和 eQTL。通过模拟,我们表明 bMIND 提高了样本水平 CTS 表达估计的准确性,并与现有方法相比,提高了发现 CTS DEG 的能力。为了进一步了解两种复杂表型,自闭症谱系障碍和阿尔茨海默病,我们将 bMIND 应用于相关脑组织的基因表达数据,以识别 CTS DEG。我们的结果补充了 snRNA-seq 研究中获得的 CTS DEG 结果,在特定细胞类型中复制了某些 DEG,同时为这些细胞类型提名了其他新基因。最后,我们通过分析基因型组织表达项目数据计算了 11 个大脑区域的 CTS eQTL,为生物学见解创造了新的资源。