Department of Microbiology, Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University, Wuhan, HB, China.
Department of Microbiology, Molecular Genetics and Immunology, University of Kansas Medical Center, Kansas City, KS, United States of America.
PLoS Genet. 2021 Apr 15;17(4):e1009366. doi: 10.1371/journal.pgen.1009366. eCollection 2021 Apr.
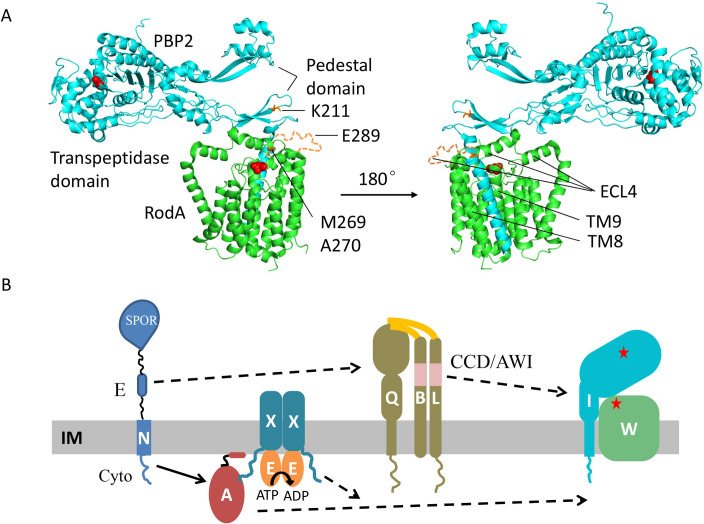
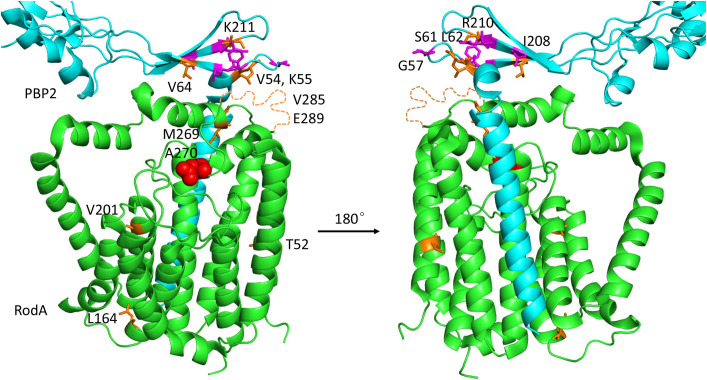
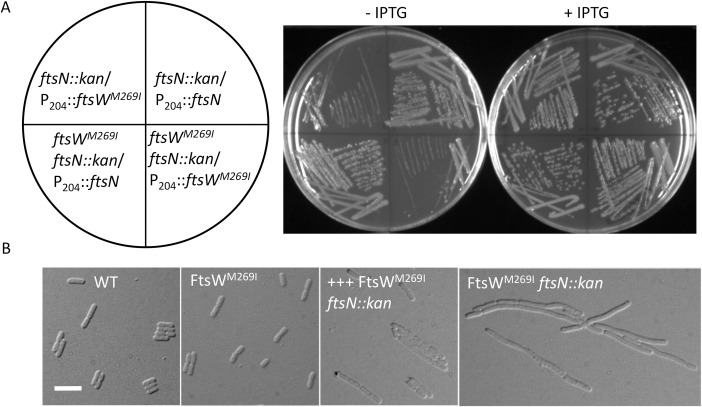
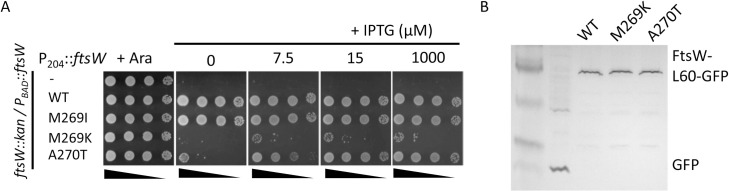
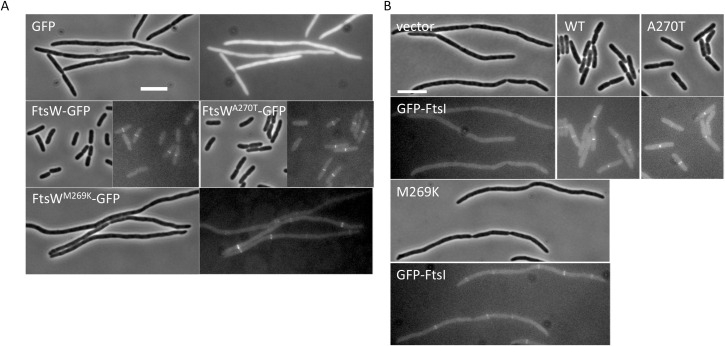
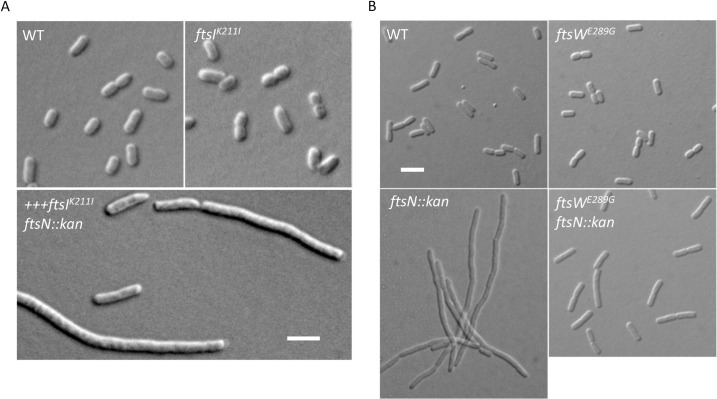
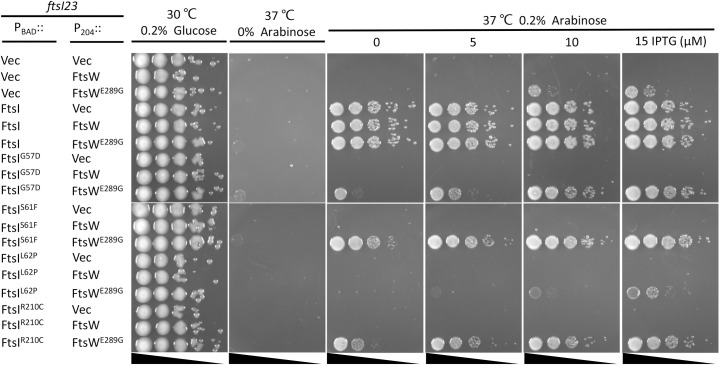
SEDS family peptidoglycan (PG) glycosyltransferases, RodA and FtsW, require their cognate transpeptidases PBP2 and FtsI (class B penicillin binding proteins) to synthesize PG along the cell cylinder and at the septum, respectively. The activities of these SEDS-bPBPs complexes are tightly regulated to ensure proper cell elongation and division. In Escherichia coli FtsN switches FtsA and FtsQLB to the active forms that synergize to stimulate FtsWI, but the exact mechanism is not well understood. Previously, we isolated an activation mutation in ftsW (M269I) that allows cell division with reduced FtsN function. To try to understand the basis for activation we isolated additional substitutions at this position and found that only the original substitution produced an active mutant whereas drastic changes resulted in an inactive mutant. In another approach we isolated suppressors of an inactive FtsL mutant and obtained FtsWE289G and FtsIK211I and found they bypassed FtsN. Epistatic analysis of these mutations and others confirmed that the FtsN-triggered activation signal goes from FtsQLB to FtsI to FtsW. Mapping these mutations, as well as others affecting the activity of FtsWI, on the RodA-PBP2 structure revealed they are located at the interaction interface between the extracellular loop 4 (ECL4) of FtsW and the pedestal domain of FtsI (PBP3). This supports a model in which the interaction between the ECL4 of SEDS proteins and the pedestal domain of their cognate bPBPs plays a critical role in the activation mechanism.
SEDS 家族肽聚糖(PG)糖基转移酶 RodA 和 FtsW 需要其同源转肽酶 PBP2 和 FtsI(B 类青霉素结合蛋白)分别在细胞柱和隔膜处合成 PG。这些 SEDS-bPBPs 复合物的活性受到严格调控,以确保细胞正常伸长和分裂。在大肠杆菌中,FtsN 将 FtsA 和 FtsQLB 切换到激活形式,协同作用刺激 FtsWI,但具体机制尚不清楚。之前,我们分离到一个 ftsW 的激活突变(M269I),该突变允许在 FtsN 功能降低的情况下进行细胞分裂。为了尝试理解激活的基础,我们在该位置分离了其他取代,发现只有原始取代产生了活性突变,而剧烈的变化则导致了无活性突变。在另一种方法中,我们分离了一个无活性 FtsL 突变体的抑制子,并获得了 FtsWE289G 和 FtsIK211I,发现它们绕过了 FtsN。这些突变和其他突变的上位性分析证实,FtsN 触发的激活信号从 FtsQLB 传递到 FtsI 再到 FtsW。将这些突变以及其他影响 FtsWI 活性的突变映射到 RodA-PBP2 结构上,表明它们位于 FtsW 的细胞外环 4(ECL4)和 FtsI 的基架域(PBP3)之间的相互作用界面上。这支持了一种模型,即 SEDS 蛋白的 ECL4 与它们同源 bPBPs 的基架域之间的相互作用在激活机制中起着关键作用。