Animal Health Department, NEIKER - Basque Institute for Agricultural Research and Development, Basque Research and Technology Alliance (BRTA), Bizkaia Science and Technology Park 812L, 48160, Derio, Bizkaia, Spain.
Applied Mathematics Department, Bioinformatics Unit, NEIKER - Basque Institute for Agricultural Research and Development, Basque Research and Technology Alliance (BRTA), Bizkaia Science and Technology Park 812L, 48160, Derio, Bizkaia, Spain.
Sci Rep. 2021 Apr 26;11(1):8998. doi: 10.1038/s41598-021-88318-0.
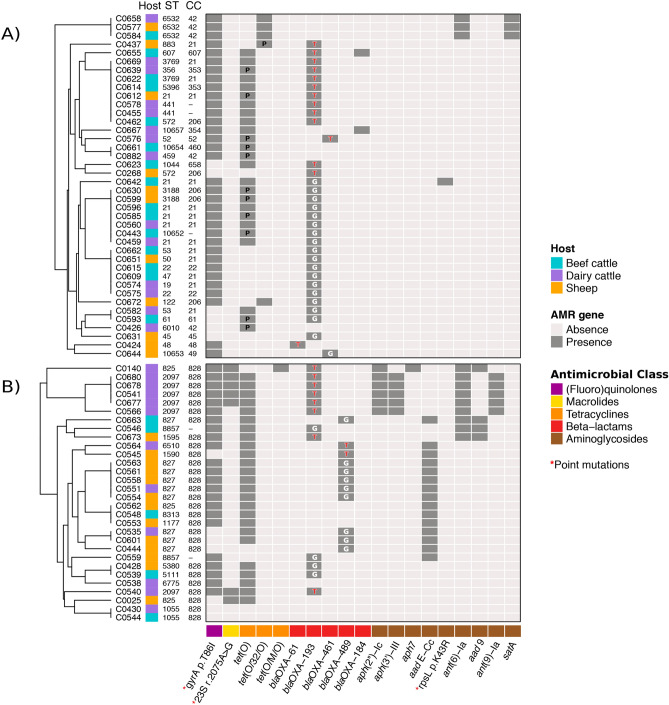
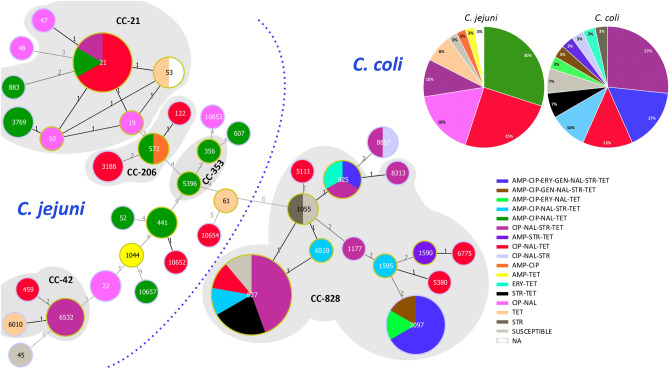
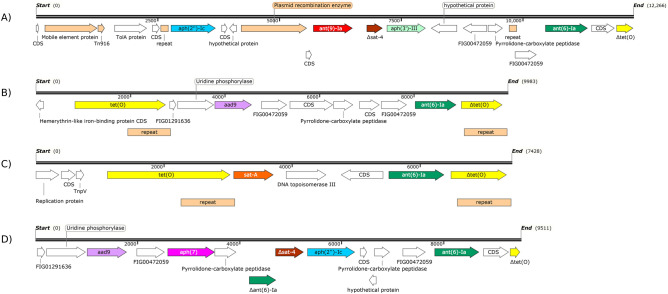
Campylobacter, a leading cause of gastroenteritis in humans, asymptomatically colonises the intestinal tract of a wide range of animals.Although antimicrobial treatment is restricted to severe cases, the increase of antimicrobial resistance (AMR) is a concern. Considering the significant contribution of ruminants as reservoirs of resistant Campylobacter, Illumina whole-genome sequencing was used to characterise the mechanisms of AMR in Campylobacter jejuni and Campylobacter coli recovered from beef cattle, dairy cattle, and sheep in northern Spain. Genome analysis showed extensive genetic diversity that clearly separated both species. Resistance genotypes were identified by screening assembled sequences with BLASTn and ABRicate, and additional sequence alignments were performed to search for frameshift mutations and gene modifications. A high correlation was observed between phenotypic resistance to a given antimicrobial and the presence of the corresponding known resistance genes. Detailed sequence analysis allowed us to detect the recently described mosaic tet(O/M/O) gene in one C. coli, describe possible new alleles of bla-like genes, and decipher the genetic context of aminoglycoside resistance genes, as well as the plasmid/chromosomal location of the different AMR genes and their implication for resistance spread. Updated resistance gene databases and detailed analysis of the matched open reading frames are needed to avoid errors when using WGS-based analysis pipelines for AMR detection in the absence of phenotypic data.
空肠弯曲菌是人类肠胃炎的主要病原体之一,无症状地定植于多种动物的肠道中。尽管抗菌治疗仅限于严重病例,但抗菌药物耐药性(AMR)的增加令人担忧。考虑到反刍动物作为耐药空肠弯曲菌的储存宿主的重要作用,本研究使用 Illumina 全基因组测序技术,对来自西班牙北部肉牛、奶牛和绵羊中分离的空肠弯曲菌和结肠弯曲菌的 AMR 机制进行了研究。基因组分析显示了广泛的遗传多样性,明确地区分了这两个种。通过 BLASTn 和 ABRicate 对组装序列进行筛选来鉴定耐药基因型,并进行额外的序列比对以搜索移码突变和基因修饰。表型对特定抗菌药物的耐药性与相应已知耐药基因的存在之间存在高度相关性。详细的序列分析使我们能够在一个结肠弯曲菌中检测到最近描述的镶嵌 tet(O/M/O)基因,描述 bla 样基因的可能新等位基因,并破译氨基糖苷类耐药基因的遗传背景,以及不同 AMR 基因的质粒/染色体位置及其对耐药性传播的影响。在没有表型数据的情况下,使用基于 WGS 的分析管道进行 AMR 检测时,需要更新耐药基因数据库并详细分析匹配的开放阅读框,以避免错误。