Institute of Biomedicine, Yliopistonranta 1E, University of Eastern Finland, 70211 Kuopio, Finland.
Structural and Computational Biology Unit, European Molecular Biology Laboratory, Meyerhofstraße 1, 69117 Heidelberg, Germany.
Cells. 2021 Apr 9;10(4):860. doi: 10.3390/cells10040860.
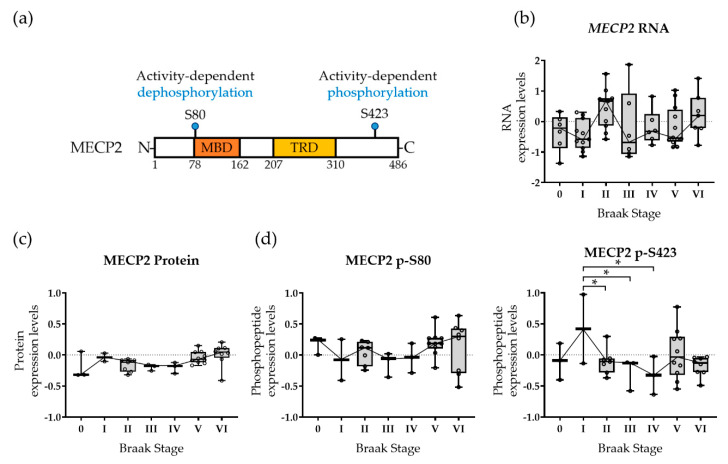
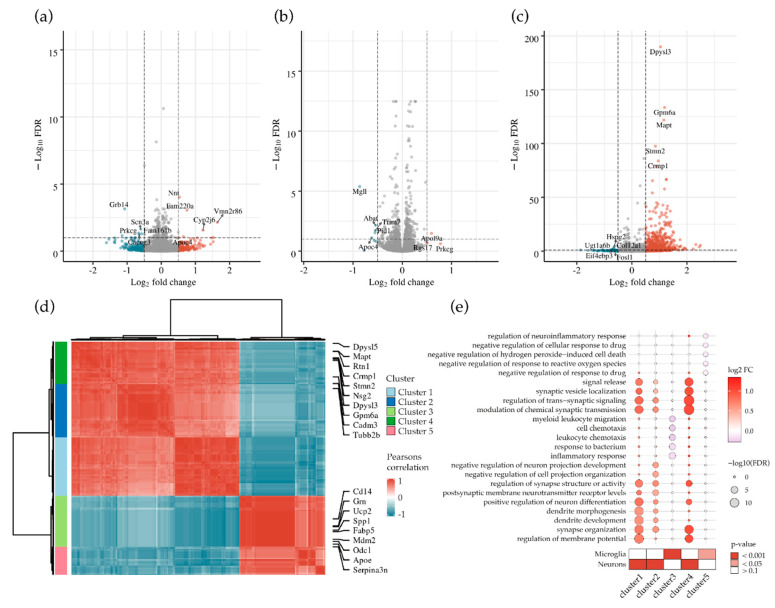
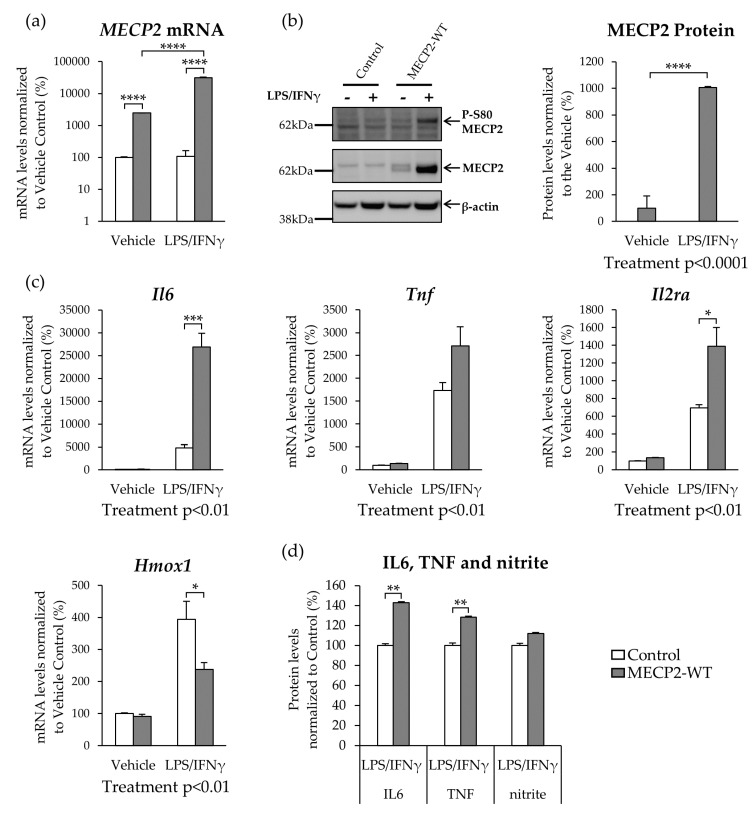
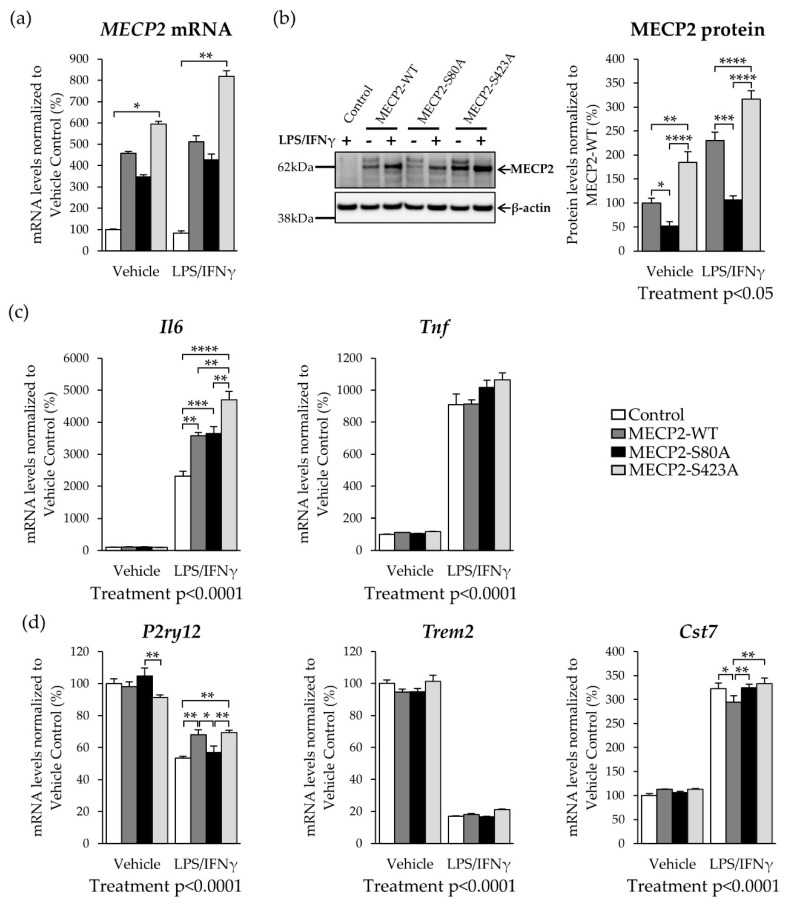
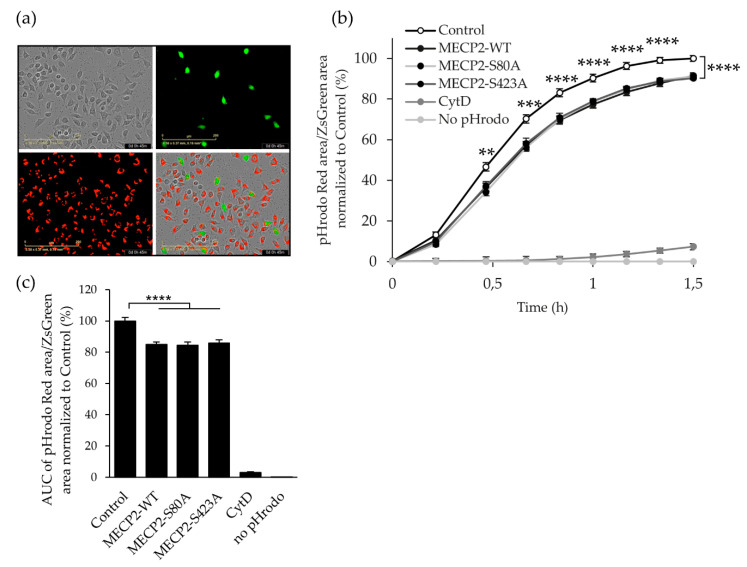
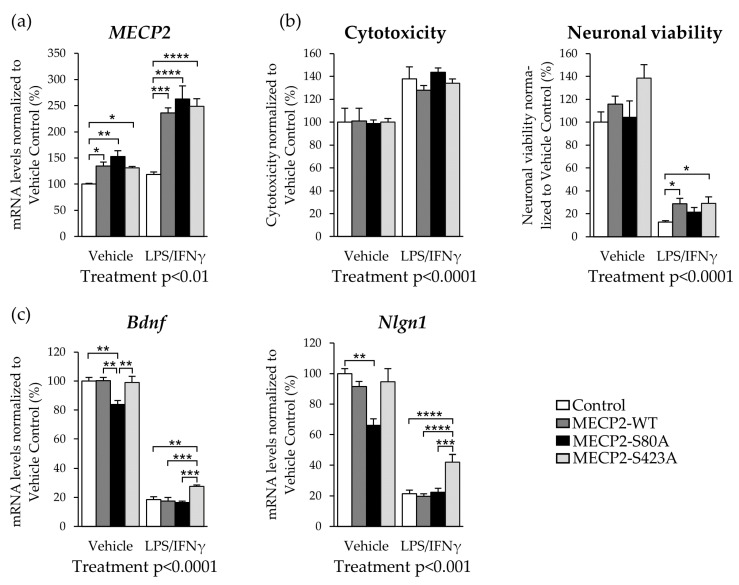
Methyl-CpG-binding protein 2 (MECP2) is a critical transcriptional regulator for synaptic function. Dysfunction of synapses, as well as microglia-mediated neuroinflammation, represent the earliest pathological events in Alzheimer's disease (AD). Here, expression, protein levels, and activity-related phosphorylation changes of MECP2 were analyzed in post-mortem human temporal cortex. The effects of wild type and phosphorylation-deficient MECP2 variants at serine 423 (S423) or S80 on microglial and neuronal function were assessed utilizing BV2 microglial monocultures and co-cultures with mouse cortical neurons under inflammatory stress conditions. MECP2 phosphorylation at the functionally relevant S423 site nominally decreased in the early stages of AD-related neurofibrillary pathology in the human temporal cortex. Overexpression of wild type MECP2 enhanced the pro-inflammatory response in BV2 cells upon treatment with lipopolysaccharide (LPS) and interferon-γ (IFNγ) and decreased BV2 cell phagocytic activity. The expression of the phosphorylation-deficient MECP2-S423A variant, but not S80A, further increased the pro-inflammatory response of BV2 cells. In neurons co-cultured with BV2 cells, the MECP2-S423A variant increased the expression of several genes, which are important for the maintenance and protection of neurons and synapses upon inflammatory stress. Collectively, functional analyses in different cellular models suggest that MECP2 may influence the inflammatory response in microglia independently of S423 and S80 phosphorylation, while the S423 phosphorylation might play a role in the activation of neuronal gene expression, which conveys neuroprotection under neuroinflammation-related stress.
甲基化 CpG 结合蛋白 2(MECP2)是突触功能的关键转录调节因子。突触功能障碍以及小胶质细胞介导的神经炎症是阿尔茨海默病(AD)最早的病理事件。在这里,分析了尸检后人类颞叶皮层中 MECP2 的表达、蛋白水平和与活性相关的磷酸化变化。利用 BV2 小胶质细胞单核培养物和在炎症应激条件下与小鼠皮质神经元共培养物,评估了野生型和磷酸化缺陷 MECP2 变体(丝氨酸 423 [S423]或 S80)对小胶质细胞和神经元功能的影响。在 AD 相关神经纤维病理的早期阶段,人类颞叶皮层中 MECP2 的功能相关 S423 位点的磷酸化程度名义上降低。在脂多糖(LPS)和干扰素-γ(IFNγ)处理下,野生型 MECP2 的过表达增强了 BV2 细胞的促炎反应,并降低了 BV2 细胞的吞噬活性。表达磷酸化缺陷 MECP2-S423A 变体,但不是 S80A,进一步增加了 BV2 细胞的促炎反应。在与 BV2 细胞共培养的神经元中,MECP2-S423A 变体增加了几个基因的表达,这些基因对神经元和突触在炎症应激下的维持和保护很重要。总的来说,不同细胞模型中的功能分析表明,MECP2 可能独立于 S423 和 S80 磷酸化影响小胶质细胞的炎症反应,而 S423 磷酸化可能在神经元基因表达的激活中发挥作用,从而在神经炎症相关应激下提供神经保护。