Ankrah Nana Y D, Barker Brandon E, Song Joan, Wu Cindy, McMullen John G, Douglas Angela E
Department of Entomology, Cornell University, Ithaca, New York, USA
Center for Advanced Computing, Cornell University, Ithaca, New York, USA.
mSystems. 2021 May 4;6(3):e01369-20. doi: 10.1128/mSystems.01369-20.
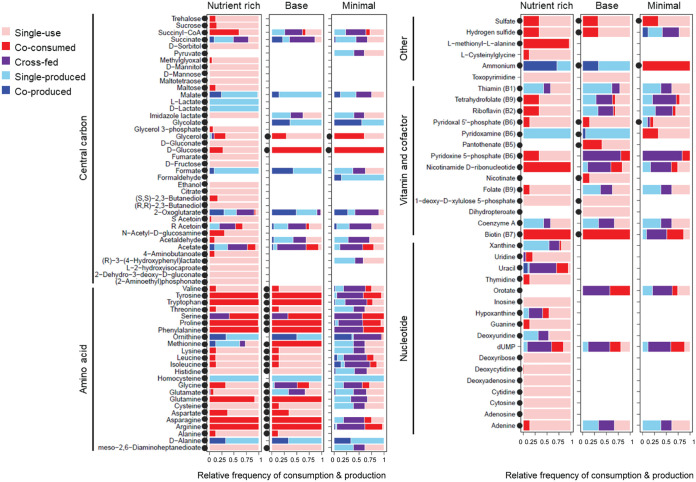
An important goal for many nutrition-based microbiome studies is to identify the metabolic function of microbes in complex microbial communities and their impact on host physiology. This research can be confounded by poorly understood effects of community composition and host diet on the metabolic traits of individual taxa. Here, we investigated these multiway interactions by constructing and analyzing metabolic models comprising every combination of five bacterial members of the gut microbiome (from single taxa to the five-member community of and species) under three nutrient regimes. We show that the metabolic function of gut bacteria is dynamic, influenced by community composition, and responsive to dietary modulation. Furthermore, we show that ecological interactions such as competition and mutualism identified from the growth patterns of gut bacteria are underlain by a diversity of metabolic interactions, and show that the bacteria tend to compete for amino acids and B vitamins more frequently than for carbon sources. Our results reveal that, in addition to fermentation products such as acetate, intermediates of the tricarboxylic acid (TCA) cycle, including 2-oxoglutarate and succinate, are produced at high flux and cross-fed between bacterial taxa, suggesting important roles for TCA cycle intermediates in modulating gut microbe interactions and the potential to influence host traits. These metabolic models provide specific predictions of the patterns of ecological and metabolic interactions among gut bacteria under different nutrient regimes, with potentially important consequences for overall community metabolic function and nutritional interactions with the host. is an important model for microbiome research partly because of the low complexity of its mostly culturable gut microbiota. Our current understanding of how interacts with its gut microbes and how these interactions influence host traits derives almost entirely from empirical studies that focus on individual microbial taxa or classes of metabolites. These studies have failed to capture fully the complexity of metabolic interactions that occur between host and microbe. To overcome this limitation, we reconstructed and analyzed 31 metabolic models for every combination of the five principal bacterial taxa in the gut microbiome of This revealed that metabolic interactions between gut bacterial taxa are highly dynamic and influenced by cooccurring bacteria and nutrient availability. Our results generate testable hypotheses about among-microbe ecological interactions in the gut and the diversity of metabolites available to influence host traits.
许多基于营养的微生物组研究的一个重要目标是确定复杂微生物群落中微生物的代谢功能及其对宿主生理的影响。这项研究可能会因群落组成和宿主饮食对单个分类群代谢特征的影响了解不足而受到干扰。在这里,我们通过构建和分析代谢模型来研究这些多向相互作用,这些模型包含肠道微生物组的五个细菌成员的每种组合(从单个分类群到和物种的五成员群落),处于三种营养状态下。我们表明,肠道细菌的代谢功能是动态的,受群落组成影响,并对饮食调节有反应。此外,我们表明,从肠道细菌生长模式中识别出的竞争和互利共生等生态相互作用是由多种代谢相互作用支撑的,并且表明细菌比争夺碳源更频繁地争夺氨基酸和B族维生素。我们的结果表明,除了乙酸盐等发酵产物外,三羧酸(TCA)循环的中间体,包括2-氧代戊二酸和琥珀酸,以高通量产生并在细菌分类群之间交叉供应,这表明TCA循环中间体在调节肠道微生物相互作用以及影响宿主特征方面具有重要作用。这些代谢模型提供了不同营养状态下肠道细菌之间生态和代谢相互作用模式的具体预测,对整体群落代谢功能以及与宿主的营养相互作用可能产生重要影响。是微生物组研究的一个重要模型,部分原因是其大部分可培养的肠道微生物群复杂性较低。我们目前对与肠道微生物如何相互作用以及这些相互作用如何影响宿主特征的理解几乎完全来自于专注于单个微生物分类群或代谢物类别的实证研究。这些研究未能充分捕捉宿主与微生物之间发生的代谢相互作用的复杂性。为了克服这一限制,我们为肠道微生物组中五个主要细菌分类群的每种组合重建并分析了31个代谢模型。这表明肠道细菌分类群之间的代谢相互作用高度动态,受共存细菌和营养可用性的影响。我们的结果产生了关于肠道中微生物间生态相互作用以及可影响宿主特征的代谢物多样性的可测试假设。