Schönborn Jürgen W, Stewart Fiona A, Enriquez Kerstin Maas, Akhtar Irfan, Droste Andrea, Waschina Silvio, Beller Mathias
Institut für Mathematische Modellierung Biologischer Systeme, Heinrich-Heine-Universität Düsseldorf, 40225 Düsseldorf, Germany.
Systembiologie des Fettstoffwechsels, Heinrich-Heine-Universität Düsseldorf, 40225 Düsseldorf, Germany.
iScience. 2021 Oct 6;24(11):103216. doi: 10.1016/j.isci.2021.103216. eCollection 2021 Nov 19.
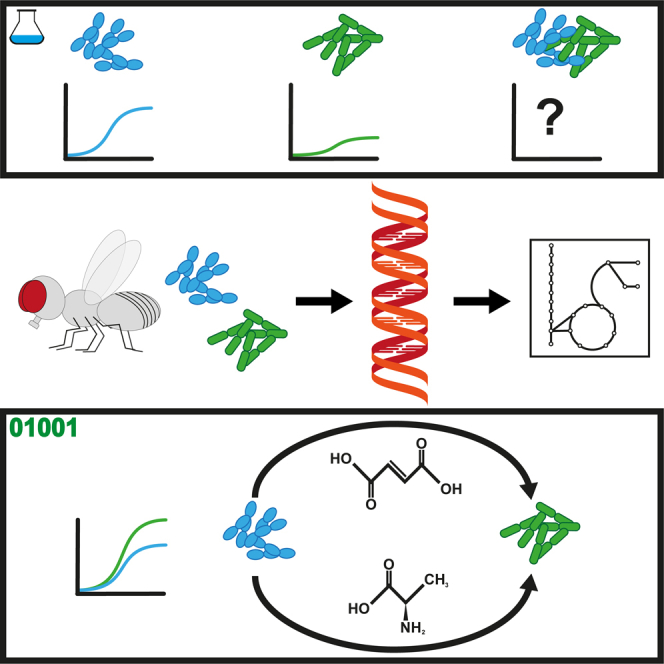
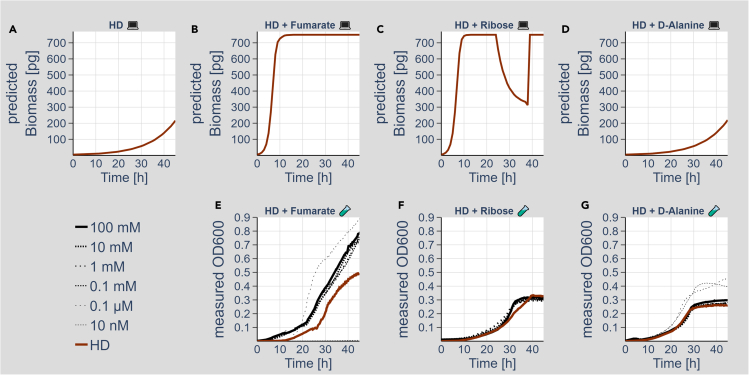
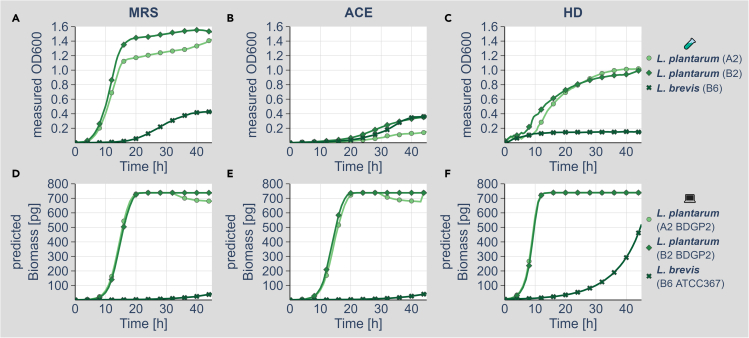
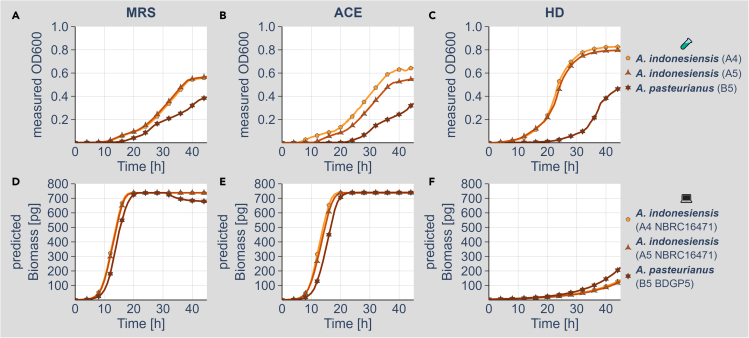
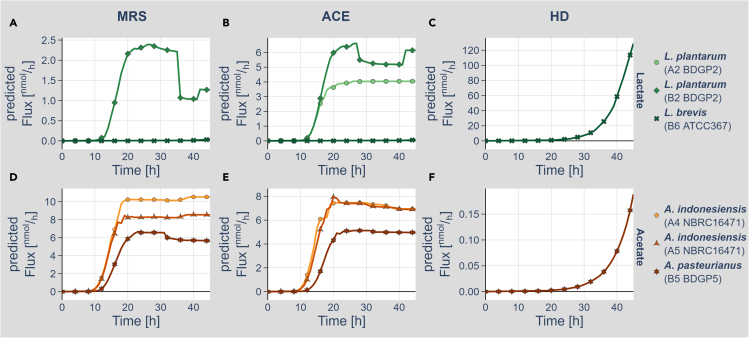
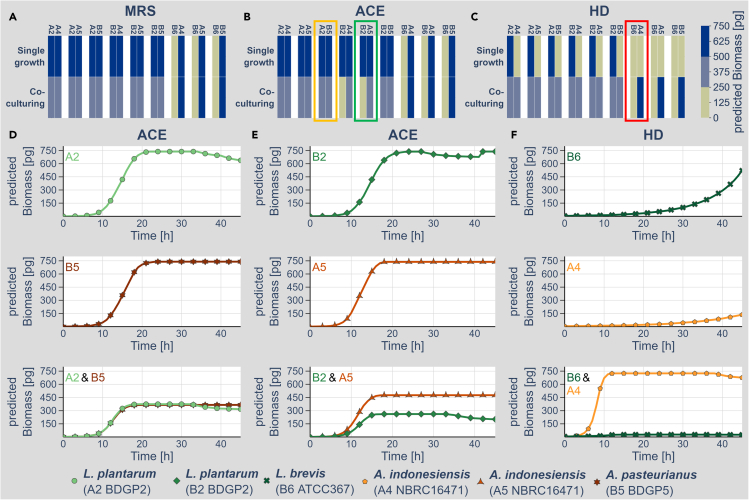
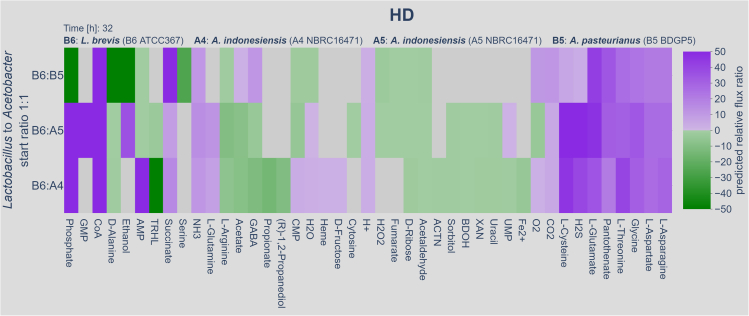
We know a lot about varying gut microbiome compositions. Yet, how the bacteria affect each other remains elusive. In mammals, this is largely based on the sheer complexity of the microbiome with at least hundreds of different species. Thus, model organisms such as are commonly used to investigate mechanistic questions as the microbiome consists of only about 10 leading bacterial species. Here, we isolated gut bacteria from laboratory-reared , sequenced their respective genomes, and used this information to reconstruct genome-scale metabolic models. With these, we simulated growth in mono- and co-culture conditions and different media including a synthetic diet designed to grow . Our simulations reveal a synergistic growth of some but not all gut microbiome members, which stems on the exchange of distinct metabolites including tricarboxylic acid cycle intermediates. Culturing experiments confirmed our predictions. Our study thus demonstrates the possibility to predict microbiome-derived growth-promoting cross-feeding.
我们对肠道微生物群组成的多样性了解很多。然而,细菌之间如何相互影响仍然难以捉摸。在哺乳动物中,这在很大程度上是由于微生物群的极度复杂性,其中至少有数百种不同的物种。因此,诸如[具体物种名称缺失]等模式生物通常被用于研究机制问题,因为其微生物群仅由大约10种主要细菌物种组成。在这里,我们从实验室饲养的[具体物种名称缺失]中分离出肠道细菌,对它们各自的基因组进行测序,并利用这些信息重建基因组规模的代谢模型。利用这些模型,我们模拟了单培养和共培养条件下以及不同培养基中的生长情况,包括一种设计用于培养[具体物种名称缺失]的合成饮食。我们的模拟揭示了一些但并非所有肠道微生物群成员的协同生长,这源于包括三羧酸循环中间体在内的不同代谢物的交换。培养实验证实了我们的预测。因此,我们的研究证明了预测微生物群衍生的促进生长的交叉喂养的可能性。