Instituto Andaluz de Ciencias de la Tierra, CSIC-University of Granada, Av. de las Palmeras 4, 18100 Armilla, Granada, Spain.
MSME, Univ Gustave Eiffel, CNRS UMR 8208, Univ Paris-Est Créteil 8208, F-77454 Marne-la-Vallée, France.
J Chem Theory Comput. 2021 Jun 8;17(6):3571-3582. doi: 10.1021/acs.jctc.0c01083. Epub 2021 May 11.
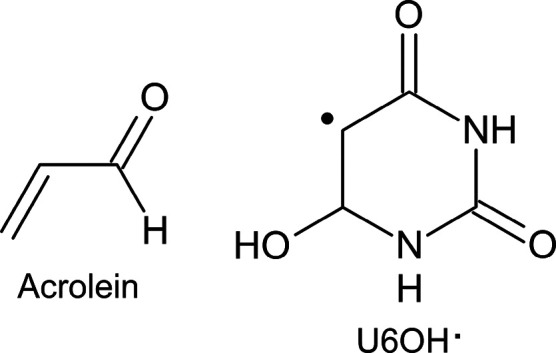
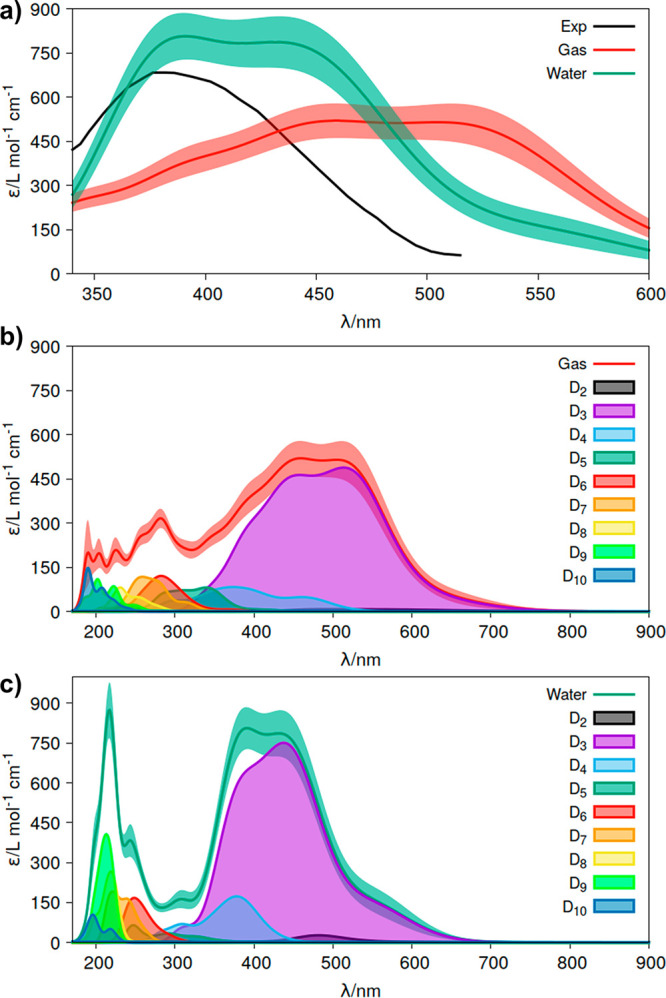
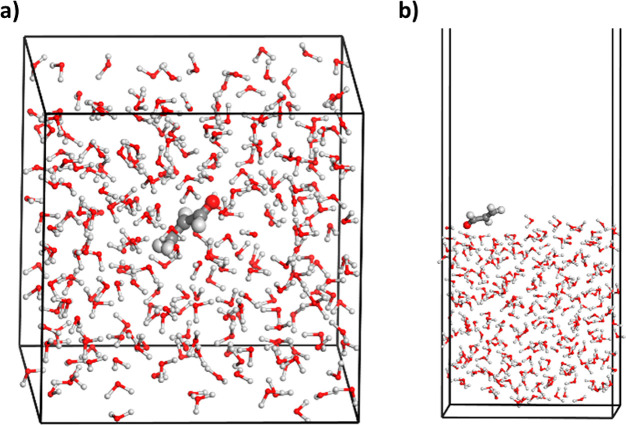
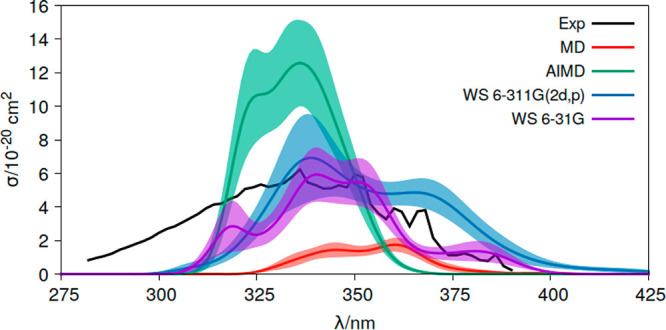
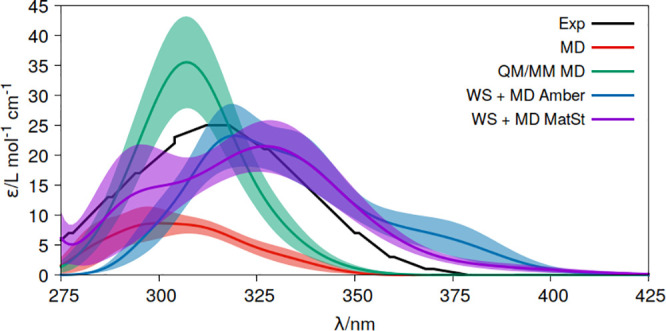
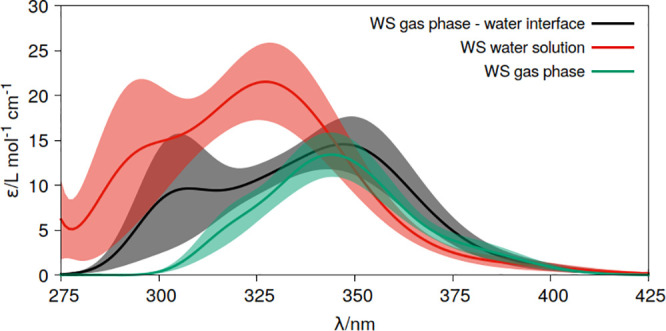
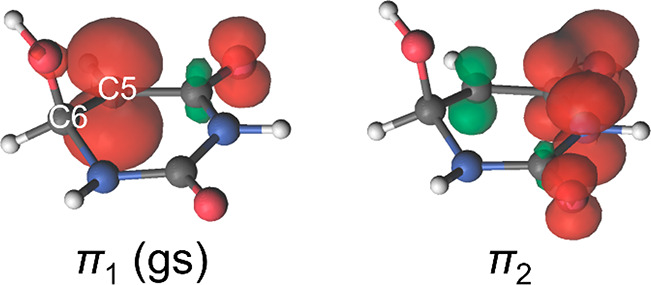
Theoretical determinations of absorption cross sections (σ) in the gas phase and molar extinction coefficients (ε) in condensed phases (water solution, interfaces or surfaces, protein or nucleic acids embeddings, etc.) are of interest when rates of photochemical processes, = ∫ ϕ(λ) σ(λ) (λ) dλ, are needed, where ϕ(λ) and (λ) are the quantum yield of the process and the irradiance of the light source, respectively, as functions of the wavelength λ. Efficient computational strategies based on single-reference quantum-chemistry methods have been developed enabling determinations of line shapes or, in some cases, achieving rovibrational resolution. Developments are however lacking for strongly correlated problems, with many excited states, high-order excitations, and/or near degeneracies between states of the same and different spin multiplicities. In this work, we define and compare the performance of distinct computational strategies using multiconfigurational quantum chemistry, nuclear sampling of the chromophore (by means of molecular dynamics, ab initio molecular dynamics, or Wigner sampling), and conformational and statistical sampling of the environment (by means of molecular dynamics). A new mathematical approach revisiting previous absolute orientation algorithms is also developed to improve alignments of geometries. These approaches are benchmarked through the π* band of acrolein not only in the gas phase and water solution but also in a gas-phase/water interface, a common situation for instance in atmospheric chemistry. Subsequently, the best strategy is used to compute the absorption band for the adduct formed upon addition of an OH radical to the C6 position of uracil and compared with the available experimental data. Overall, quantum Wigner sampling of the chromophore with molecular dynamics sampling of the environment with CASPT2 electronic-structure determinations arise as a powerful methodology to predict meaningful σ(λ) and ε(λ) band line shapes with accurate absolute intensities.
当需要计算光化学过程的速率,即 = ∫ ϕ(λ) σ(λ) (λ) dλ,其中 ϕ(λ) 和 (λ) 分别为过程的量子产率和光源的辐照度,作为波长 λ 的函数时,气态的吸收截面(σ)和凝聚相(水溶液、界面或表面、蛋白质或核酸嵌入等)的摩尔消光系数(ε)的理论确定就很有意义。已经开发了基于单参考量子化学方法的高效计算策略,这些策略能够确定谱线形状,或者在某些情况下,实现振转分辨率。然而,对于强相关问题,如存在许多激发态、高阶激发和/或相同和不同自旋多重性状态之间的近简并,这些方法的发展还存在不足。在这项工作中,我们使用多组态量子化学、发色团的核采样(通过分子动力学、从头算分子动力学或维格纳采样)以及环境的构象和统计采样(通过分子动力学)定义并比较了不同计算策略的性能。还开发了一种新的数学方法来重新审视以前的绝对取向算法,以改进几何形状的对准。通过丙烯醛的π*带,这些方法在气相和水溶液中,以及在气相/水界面(例如在大气化学中常见的情况)中进行了基准测试。随后,使用最佳策略计算了添加 OH 自由基到尿嘧啶的 C6 位形成的加合物的吸收带,并与可用的实验数据进行了比较。总体而言,发色团的量子维格纳采样与环境的分子动力学采样以及 CASPT2 电子结构测定相结合,是一种预测具有准确绝对强度的有意义的 σ(λ) 和 ε(λ) 带谱线形状的强大方法。