Coriell Institute for Medical Research, 403 Haddon Ave., Camden, NJ, 08103, USA.
Fox Chase Cancer Center, Philadelphia, PA, USA.
Breast Cancer Res. 2021 May 22;23(1):58. doi: 10.1186/s13058-021-01434-7.
DNA methylation alterations have similar patterns in normal aging tissue and in cancer. In this study, we investigated breast tissue-specific age-related DNA methylation alterations and used those methylation sites to identify individuals with outlier phenotypes. Outlier phenotype is identified by unsupervised anomaly detection algorithms and is defined by individuals who have normal tissue age-dependent DNA methylation levels that vary dramatically from the population mean.
We generated whole-genome DNA methylation profiles (GSE160233) on purified epithelial cells and used publicly available Infinium HumanMethylation 450K array datasets (TCGA, GSE88883, GSE69914, GSE101961, and GSE74214) for discovery and validation.
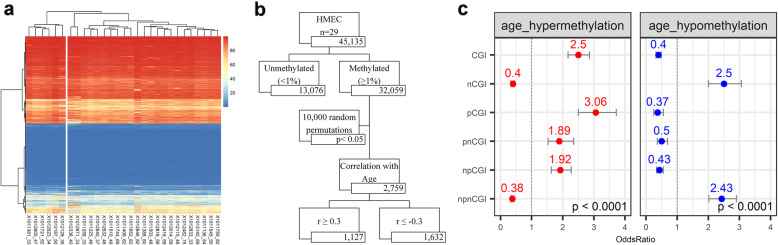
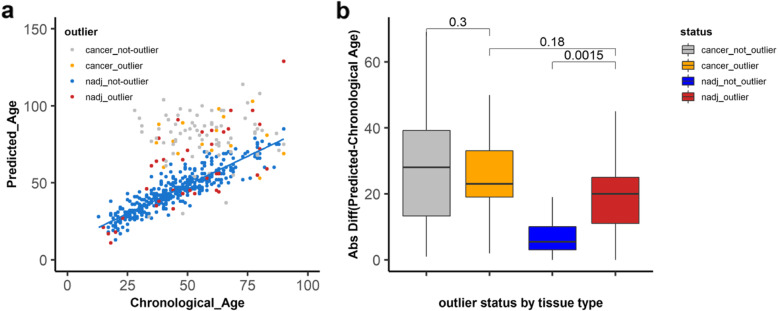
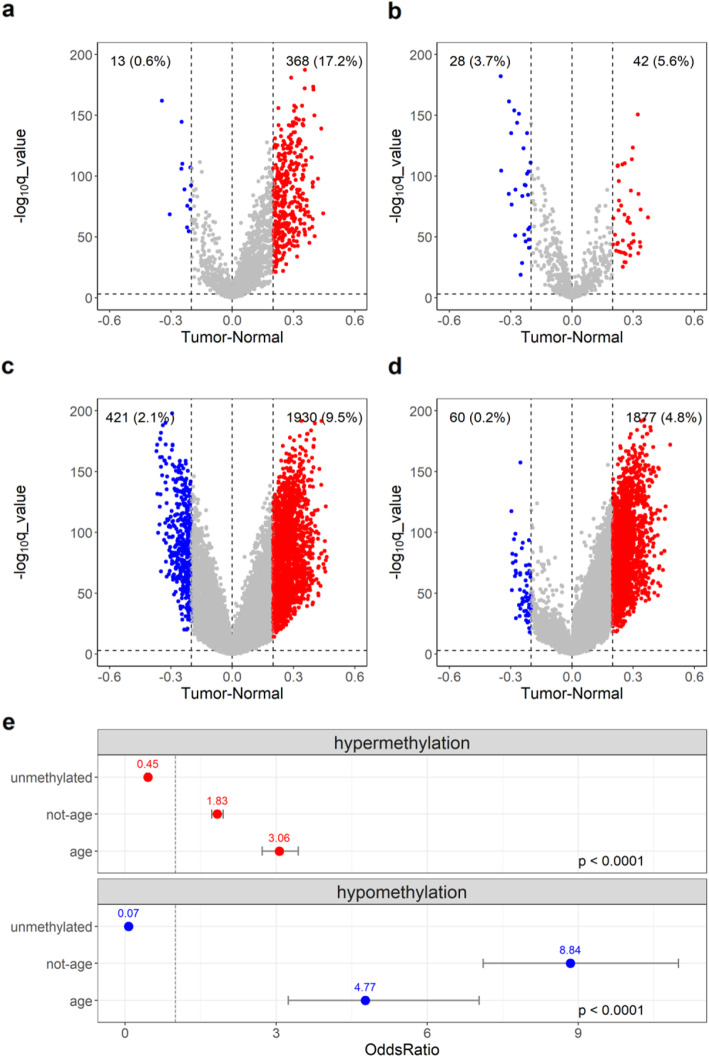
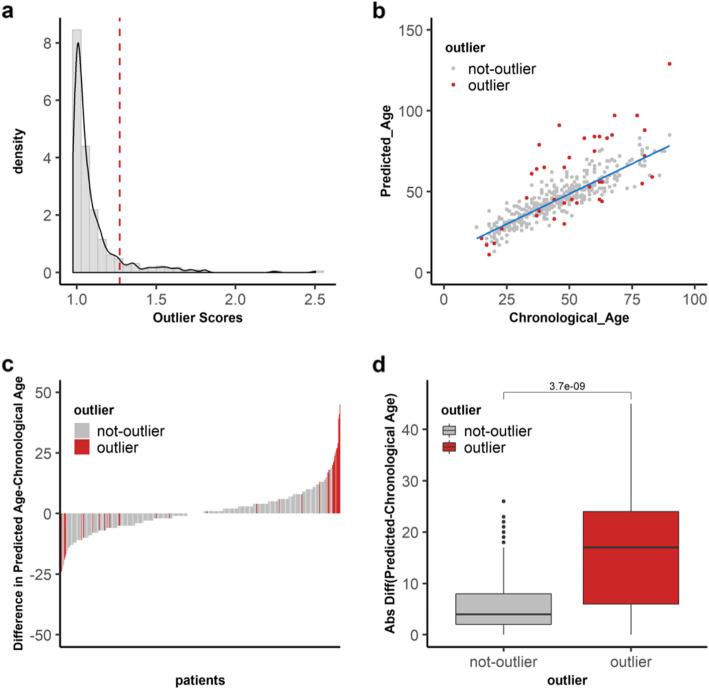
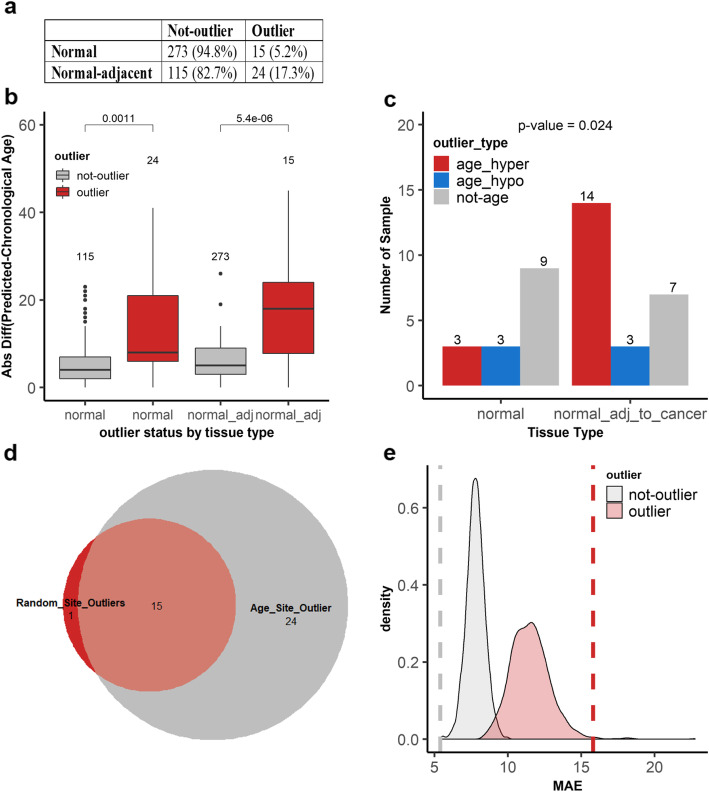
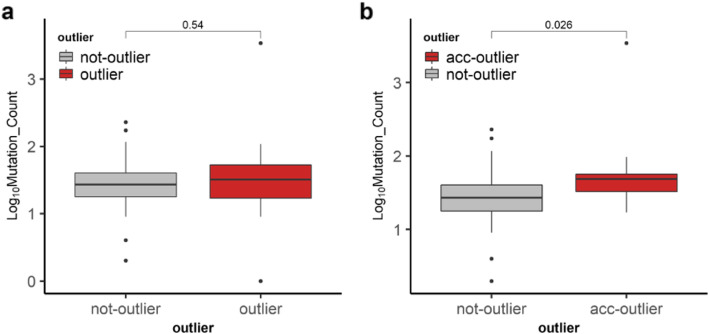
We found that hypermethylation in normal breast tissue is the best predictor of hypermethylation in cancer. Using unsupervised anomaly detection approaches, we found that about 10% of the individuals (39/427) were outliers for DNA methylation from 6 DNA methylation datasets. We also found that there were significantly more outlier samples in normal-adjacent to cancer (24/139, 17.3%) than in normal samples (15/228, 5.2%). Additionally, we found significant differences between the predicted ages based on DNA methylation and the chronological ages among outliers and not-outliers. Additionally, we found that accelerated outliers (older predicted age) were more frequent in normal-adjacent to cancer (14/17, 82%) compared to normal samples from individuals without cancer (3/17, 18%). Furthermore, in matched samples, we found that the epigenome of the outliers in the pre-malignant tissue was as severely altered as in cancer.
A subset of patients with breast cancer has severely altered epigenomes which are characterized by accelerated aging in their normal-appearing tissue. In the future, these DNA methylation sites should be studied further such as in cell-free DNA to determine their potential use as biomarkers for early detection of malignant transformation and preventive intervention in breast cancer.
在正常衰老组织和癌症中,DNA 甲基化改变具有相似的模式。在这项研究中,我们研究了乳腺组织特异性的与年龄相关的 DNA 甲基化改变,并利用这些甲基化位点来识别具有异常表型的个体。异常表型是通过无监督异常检测算法识别的,定义为具有正常组织年龄相关 DNA 甲基化水平的个体,这些水平与群体平均值有很大差异。
我们生成了纯化上皮细胞的全基因组 DNA 甲基化图谱(GSE160233),并使用了公开的 Infinium HumanMethylation 450K 阵列数据集(TCGA、GSE88883、GSE69914、GSE101961 和 GSE74214)进行发现和验证。
我们发现正常乳腺组织中的高甲基化是癌症中高甲基化的最佳预测因子。使用无监督异常检测方法,我们在 6 个 DNA 甲基化数据集中发现,约 10%的个体(39/427)的 DNA 甲基化是异常的。我们还发现,在正常-癌旁组织中异常样本的数量明显多于正常样本(24/139,17.3%)。此外,我们发现异常样本和非异常样本之间基于 DNA 甲基化预测的年龄与实际年龄之间存在显著差异。此外,我们发现,在正常-癌旁组织中,加速异常(预测年龄较大)的比例明显高于无癌症个体的正常样本(14/17,82%)。此外,在匹配样本中,我们发现癌前组织中异常样本的表观基因组改变与癌症一样严重。
乳腺癌患者的一部分具有严重改变的表观基因组,其特征是正常外观组织的加速衰老。在未来,这些 DNA 甲基化位点应该进一步研究,例如在游离 DNA 中,以确定它们作为恶性转化早期检测和乳腺癌预防干预的潜在生物标志物的用途。