Department of Biochemistry and Molecular Biology, Dalhousie University, PO Box 15000, Sir Charles Tupper Medical Building, 5850 College Street, Halifax, Nova Scotia, B3H 4R2, Canada.
Centre for Comparative Genomics and Evolutionary Bioinformatics, Dalhousie University, Halifax, Nova Scotia, Canada.
BMC Genomics. 2021 May 24;22(1):379. doi: 10.1186/s12864-021-07666-3.
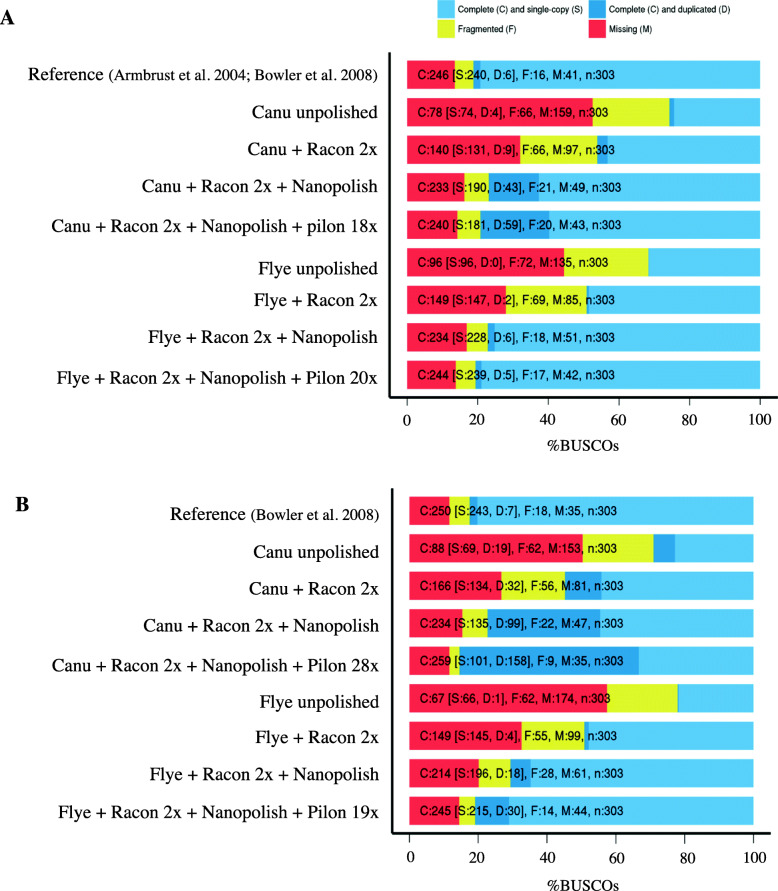
The marine diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum are valuable model organisms for exploring the evolution, diversity and ecology of this important algal group. Their reference genomes, published in 2004 and 2008, respectively, were the product of traditional Sanger sequencing. In the case of T. pseudonana, optical restriction site mapping was employed to further clarify and contextualize chromosome-level scaffolds. While both genomes are considered highly accurate and reasonably contiguous, they still contain many unresolved regions and unordered/unlinked scaffolds.
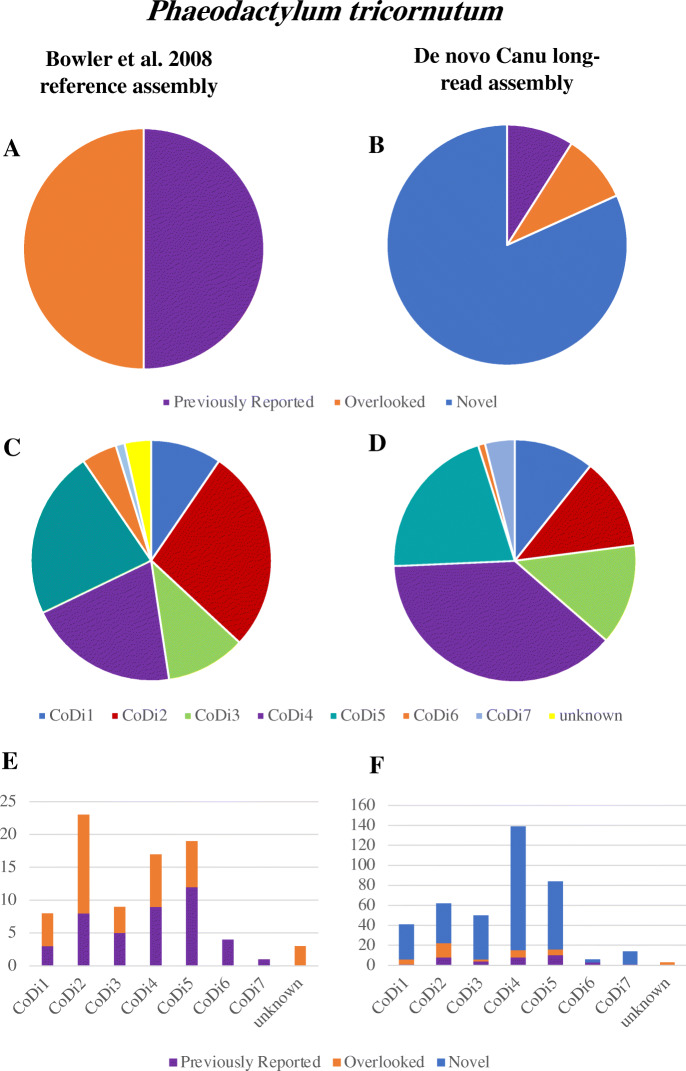
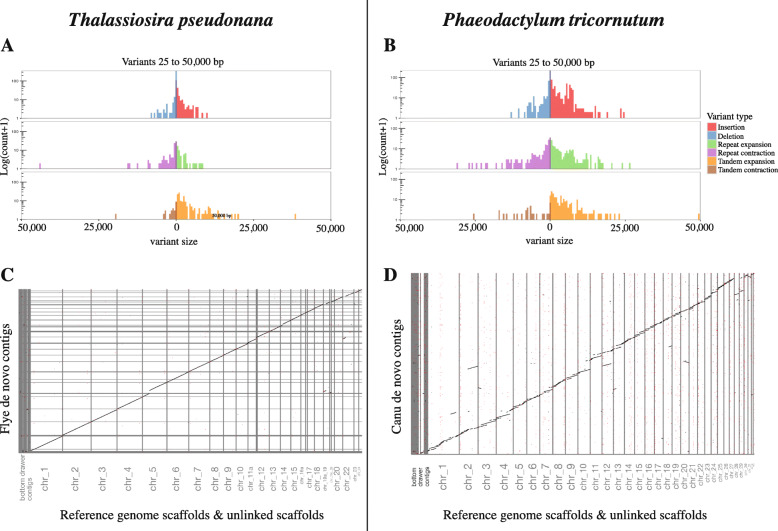
We have used Oxford Nanopore Technologies long-read sequencing to update and validate the quality and contiguity of the T. pseudonana and P. tricornutum genomes. Fine-scale assessment of our long-read derived genome assemblies allowed us to resolve previously uncertain genomic regions, further characterize complex structural variation, and re-evaluate the repetitive DNA content of both genomes. We also identified 1862 previously undescribed genes in T. pseudonana. In P. tricornutum, we used transposable element detection software to identify 33 novel copia-type LTR-RT insertions, indicating ongoing activity and rapid expansion of this superfamily as the organism continues to be maintained in culture. Finally, Bionano optical mapping of P. tricornutum chromosomes was combined with long-read sequence data to explore the potential of long-read sequencing and optical mapping for resolving haplotypes.
Despite its potential to yield highly contiguous scaffolds, long-read sequencing is not a panacea. Even for relatively small nuclear genomes such as those investigated herein, repetitive DNA sequences cause problems for current genome assembly algorithms. Determining whether a long-read derived genomic assembly is 'better' than one produced using traditional sequence data is not straightforward. Our revised reference genomes for P. tricornutum and T. pseudonana nevertheless provide additional insight into the structure and evolution of both genomes, thereby providing a more robust foundation for future diatom research.
海洋硅藻拟菱形藻和三角褐指藻是探索这一重要藻类群体进化、多样性和生态学的有价值的模式生物。它们的参考基因组分别于 2004 年和 2008 年发表,是传统桑格测序的产物。在拟菱形藻的情况下,采用光学限制位点作图进一步阐明和上下文化染色体水平支架。虽然这两个基因组被认为具有高度准确性和相当的连续性,但它们仍然包含许多未解决的区域和未排序/未链接的支架。
我们使用牛津纳米孔技术长读测序来更新和验证拟菱形藻和三角褐指藻基因组的质量和连续性。对我们的长读衍生基因组组装的精细评估使我们能够解决以前不确定的基因组区域,进一步描述复杂的结构变异,并重新评估这两个基因组的重复 DNA 含量。我们还在拟菱形藻中鉴定了 1862 个以前未描述的基因。在三角褐指藻中,我们使用转座元件检测软件鉴定了 33 个新的 copia 型 LTR-RT 插入,表明该超家族在该生物继续在培养中维持时仍在活跃和快速扩张。最后,三角褐指藻染色体的 Bionano 光学作图与长读序列数据相结合,探索了长读测序和光学作图用于解析单倍型的潜力。
尽管长读测序有可能产生高度连续的支架,但它并不是万能的。即使对于像本文所研究的相对较小的核基因组,重复 DNA 序列也会给当前的基因组组装算法带来问题。确定一个长读衍生的基因组组装是否“优于”使用传统序列数据生成的组装并不简单。然而,我们修订后的三角褐指藻和拟菱形藻参考基因组提供了对这两个基因组结构和进化的更多见解,从而为未来的硅藻研究提供了更稳健的基础。