Abdolalian Payam, Tizhoush Samaneh K, Farshadfar Kaveh, Ariafard Alireza
Department of Chemistry, Islamic Azad University Central Tehran Branch, Poonak Tehran 1469669191 Iran.
School of Natural Sciences - Chemistry, University of Tasmania Private Bag 75 Hobart TAS 7001 Australia
Chem Sci. 2021 Apr 14;12(20):7185-7195. doi: 10.1039/d1sc01230d.
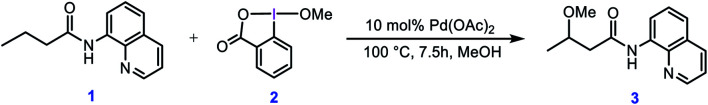
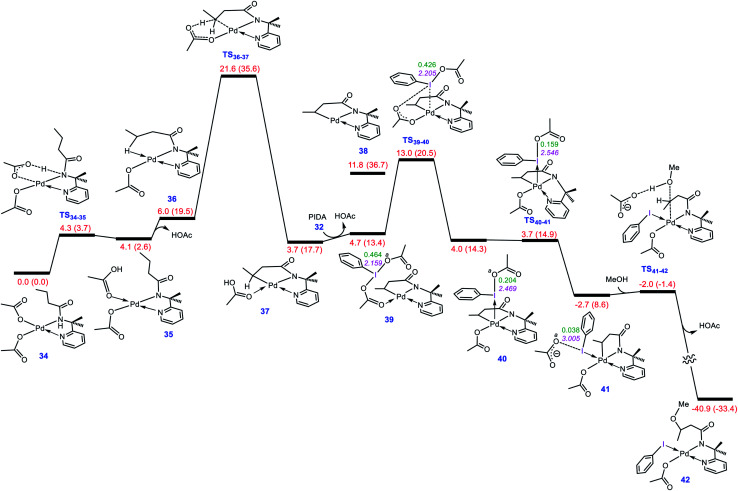
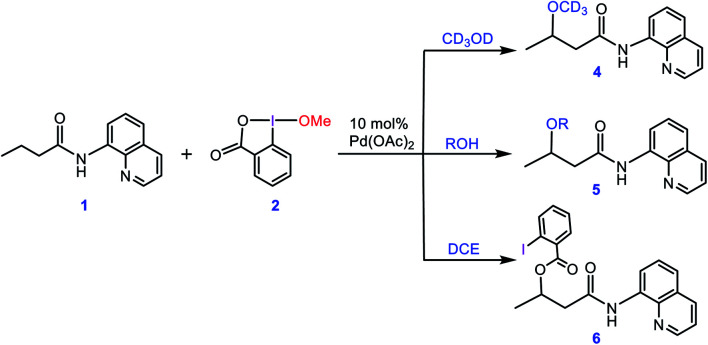
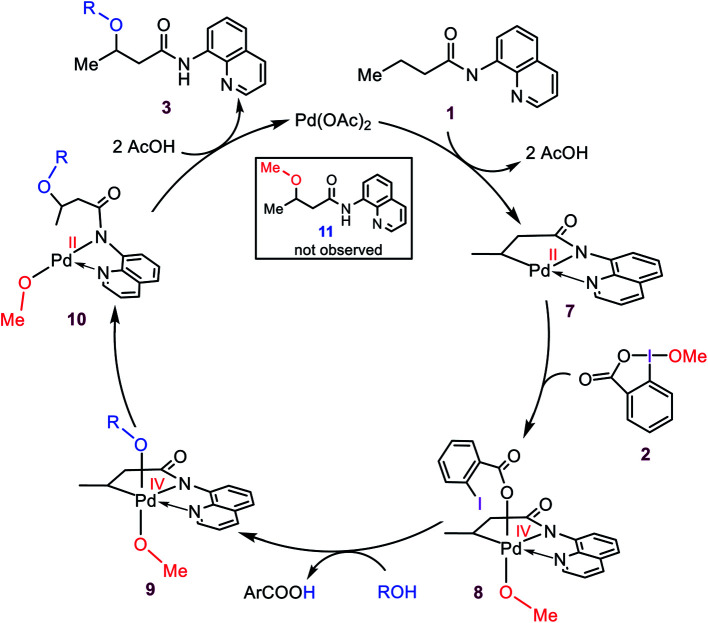
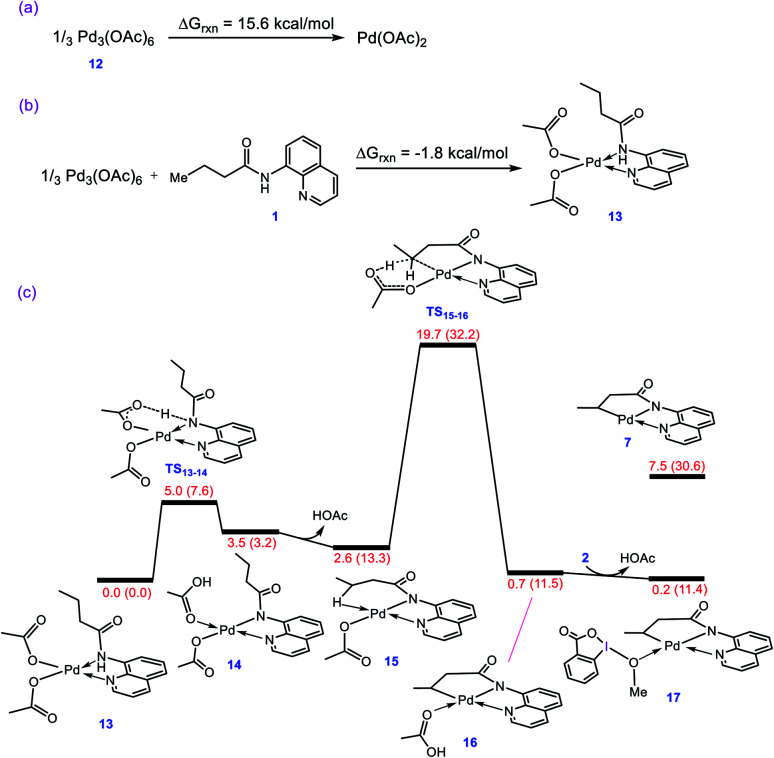
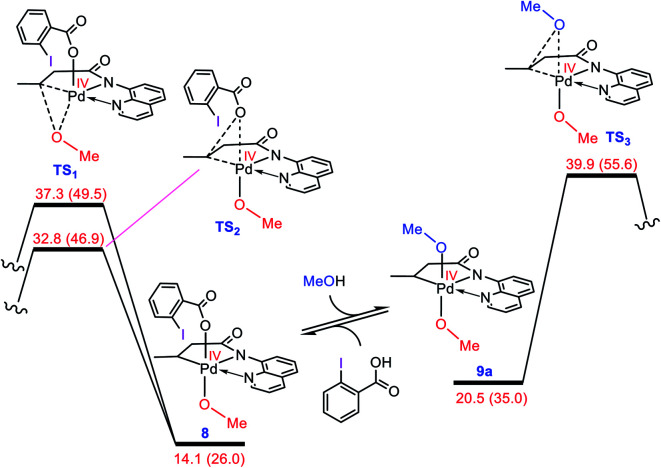
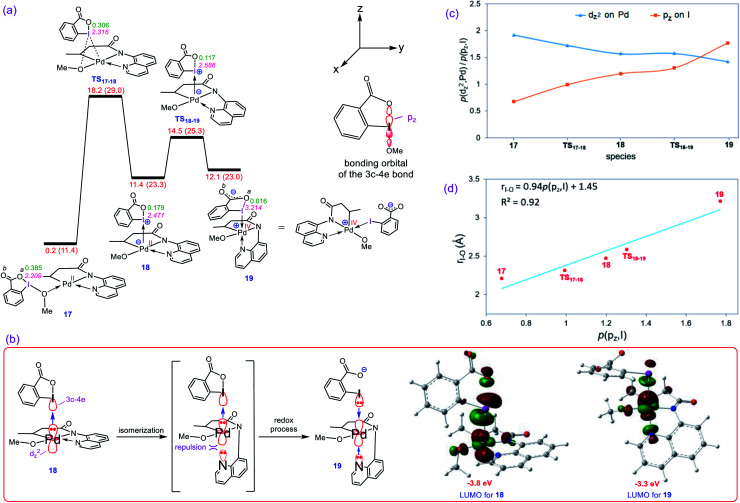
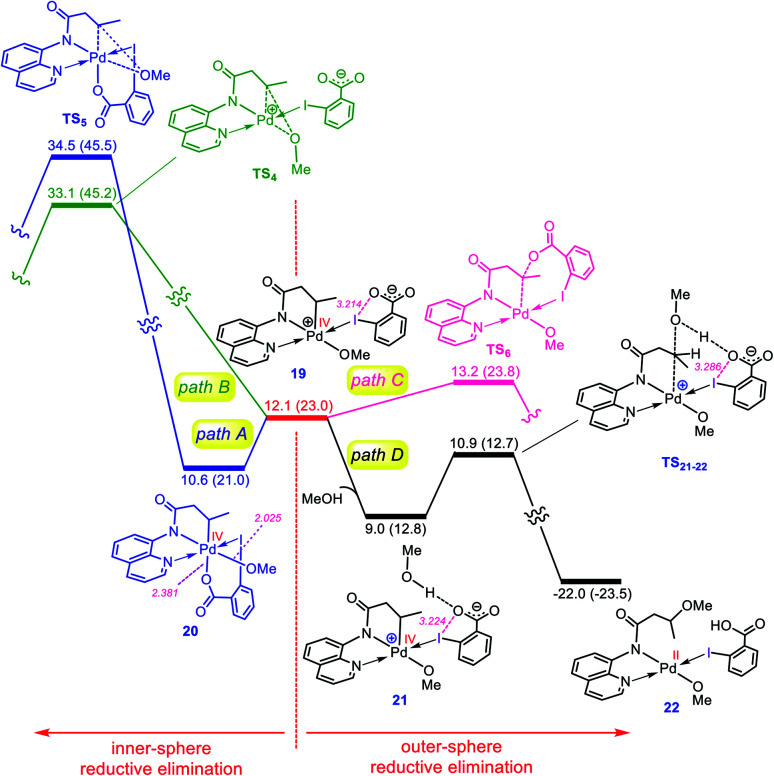
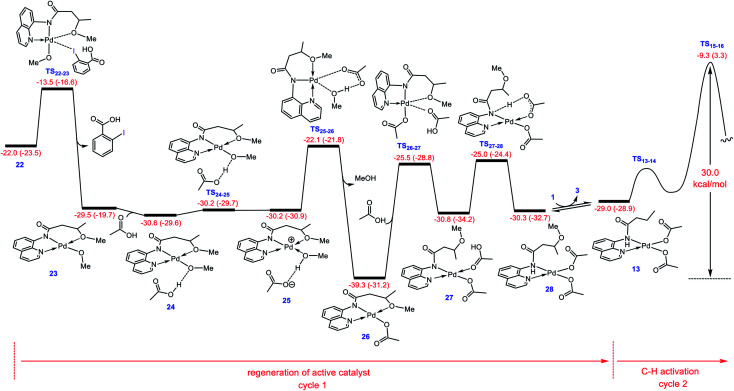
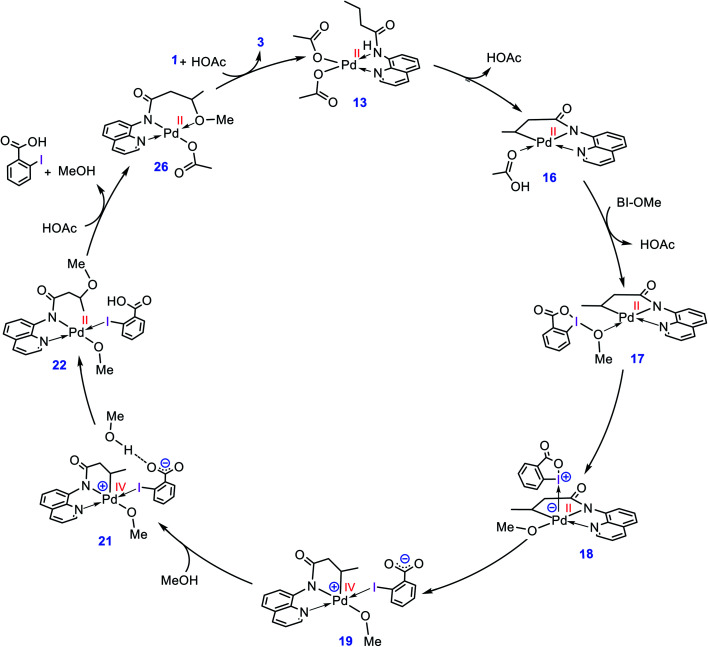
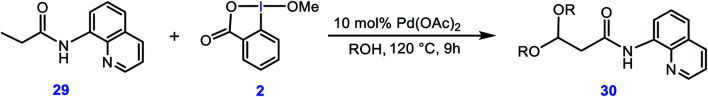
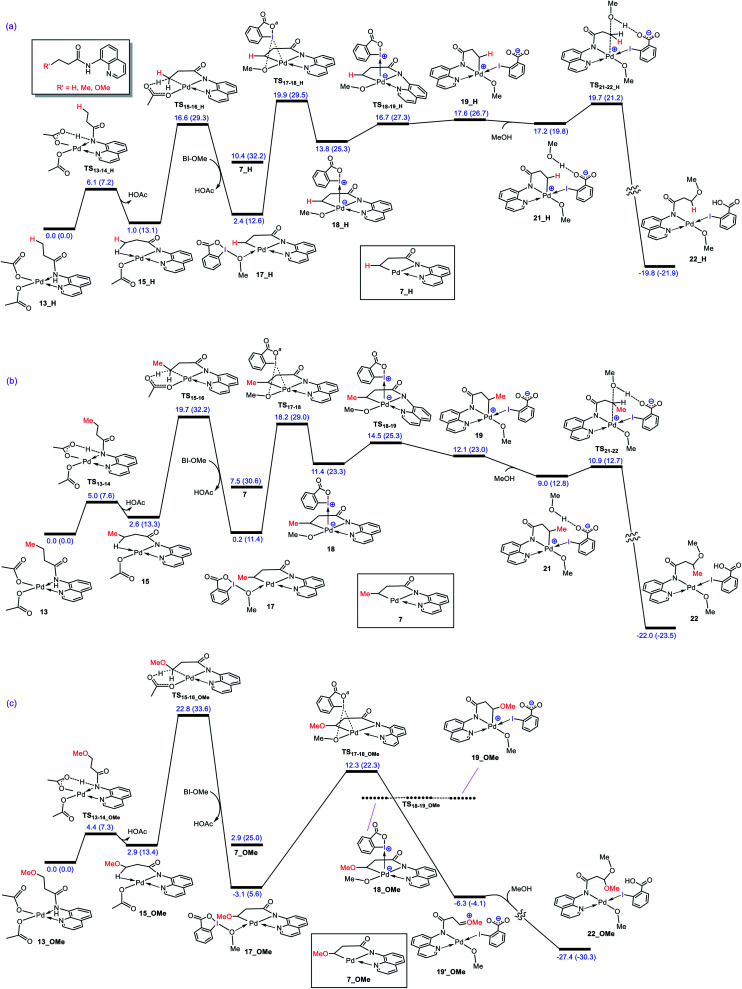
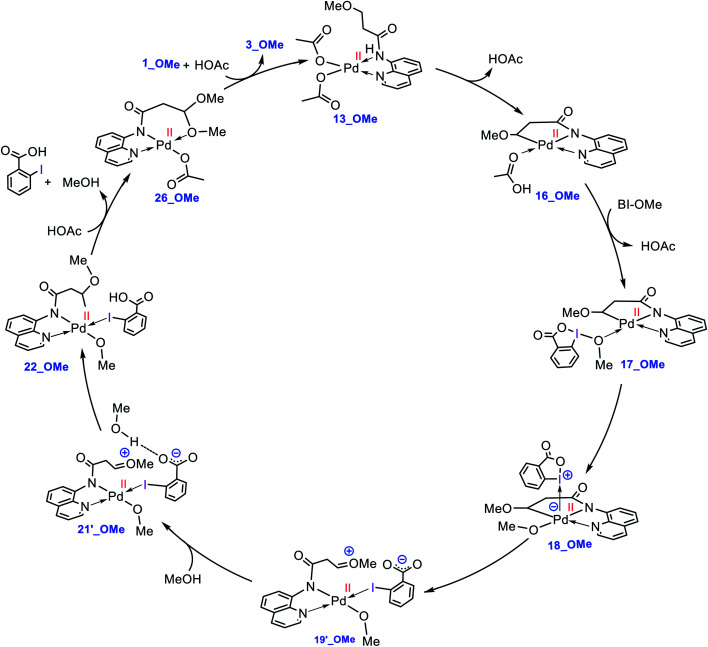
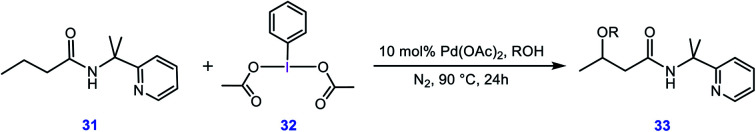
Although Pd(OAc)-catalysed alkoxylation of the C(sp)-H bonds mediated by hypervalent iodine(iii) reagents (ArIX) has been developed by several prominent researchers, there is no clear mechanism yet for such crucial transformations. In this study, we shed light on this important issue with the aid of the density functional theory (DFT) calculations for alkoxylation of butyramide derivatives. We found that the previously proposed mechanism in the literature is not consistent with the experimental observations and thus cannot be operating. The calculations allowed us to discover an unprecedented mechanism composed of four main steps as follows: (i) activation of the C(sp)-H bond, (ii) oxidative addition, (iii) reductive elimination and (iv) regeneration of the active catalyst. After completion of step (i) the CMD mechanism, the oxidative addition commences with an X ligand transfer from the iodine(iii) reagent (ArIX) to Pd(ii) to form a square pyramidal complex in which an iodonium occupies the apical position. Interestingly, a simple isomerization of the resultant five-coordinate complex triggers the Pd(ii) oxidation. Accordingly, the movement of the ligand trans to the Pd-C(sp) bond to the apical position promotes the electron transfer from Pd(ii) to iodine(iii), resulting in the reduction of iodine(iii) concomitant with the ejection of the second X ligand as a free anion. The ensuing Pd(iv) complex then undergoes the C-O reductive elimination by nucleophilic attack of the solvent (alcohol) on the sp carbon an outer-sphere S2 mechanism assisted by the X anion. Noteworthy, starting from the five coordinate complex, the oxidative addition and reductive elimination processes occur with a very low activation barrier (Δ 0-6 kcal mol). The strong coordination of the alkoxylated product to the Pd(ii) centre causes the regeneration of the active catalyst, step (iv), to be considerably endergonic, leading to subsequent catalytic cycles to proceed with a much higher activation barrier than the first cycle. We also found that although, in most cases, the alkoxylation reactions proceed a Pd(ii)-Pd(iv)-Pd(ii) catalytic cycle, the other alternative in which the oxidation state of the Pd(ii) centre remains unchanged during the catalysis could be operative, depending on the nature of the organic substrate.
尽管几位杰出的研究人员已经开发出了由高价碘(III)试剂(ArIX)介导的Pd(OAc)催化的C(sp)-H键的烷氧基化反应,但对于这种关键的转化反应,尚未有明确的反应机理。在本研究中,我们借助密度泛函理论(DFT)对丁酰胺衍生物的烷氧基化反应进行计算,阐明了这一重要问题。我们发现,文献中先前提出的反应机理与实验观察结果不一致,因此不可能起作用。计算使我们发现了一种前所未有的反应机理,该机理由以下四个主要步骤组成:(i)C(sp)-H键的活化;(ii)氧化加成;(iii)还原消除;(iv)活性催化剂的再生。在步骤(i)即CMD机理完成后,氧化加成反应开始,一个X配体从碘(III)试剂(ArIX)转移到Pd(II)上,形成一个四方锥配合物,其中一个碘鎓占据顶端位置。有趣的是,所得五配位配合物的一个简单异构化引发了Pd(II)的氧化。相应地,与Pd-C(sp)键反式的配体移动到顶端位置,促进了电子从Pd(II)转移到碘(III),导致碘(III)还原,同时第二个X配体作为游离阴离子被逐出。随后生成的Pd(IV)配合物通过溶剂(醇)对sp碳的亲核进攻进行C-O还原消除,这是一个由X阴离子辅助的外层S2机理。值得注意的是,从五配位配合物开始,氧化加成和还原消除过程以非常低的活化能垒(Δ 0 - 6 kcal/mol)发生。烷氧基化产物与Pd(II)中心的强配位作用导致活性催化剂的再生,即步骤(iv),相当程度上是吸热的,导致后续催化循环的活化能垒比第一个循环高得多。我们还发现,尽管在大多数情况下,烷氧基化反应通过Pd(II)-Pd(IV)-Pd(II)催化循环进行,但根据有机底物的性质,另一种替代情况,即Pd(II)中心的氧化态在催化过程中保持不变,也可能起作用。